Visualizing V(D)J data
Integration with scanpy
Now that we have both 1) a pre-processed V(D)J data in Dandelion object and 2) matching AnnData object, we can start finding clones and ‘integrate’ the results. All the V(D)J (AIRR) analyses files can be saved as .tsv format so that it can be used in other tools like immcantation, immunoarch, vdjtools, etc.
The results can also be ported into the AnnData object for access to more plotting functions provided through scanpy [Wolf2018].
[1]:
import os
import dandelion as ddl
import scanpy as sc
sc.settings.verbosity = 3
ddl.set_backend("base")
# change to tutorials directory
os.chdir("dandelion_tutorial")
Read in the previously saved files
I will work with the same example from the previous section since I have the AnnData object saved and vdj table filtered.
[2]:
adata = sc.read_h5ad("adata.h5ad")
adata
[2]:
AnnData object with n_obs × n_vars = 25063 × 1309
obs: 'sample_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'gmm_pct_count_clusters_keep', 'scrublet_score', 'is_doublet', 'filter_rna', 'has_contig', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'leiden'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'chain_status_colors', 'hvg', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'sample_id_colors', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'connectivities', 'distances'
[3]:
vdj = ddl.read_h5ddl("dandelion_results_simplified.h5ddl")
vdj
[3]:
Dandelion class object with n_obs = 2339 and n_contigs = 5592
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'rearrangement_status', 'v_call_functionality', 'd_call_functionality', 'j_call_functionality', 'ambiguous', 'extra', 'clone_id'
metadata: 'clone_id', 'clone_id_rank', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
layout: layout for 2339 vertices, layout for 146 vertices
graph: networkx graph of 2339 vertices, networkx graph of 146 vertices
distances: distance matrix of shape (2339, 2339)
ddl.tl.transfer
We can sync the V(D)J data from Dandelion object to the matching AnnData object using ddl.tl.transfer function.
[4]:
ddl.tl.transfer(adata, vdj)
adata
Transferring network
finished: updated `.obs` with `.metadata`
wrote active layout to `.obsm['X_vdj']`; stashed all views in `.uns['dandelion']` ('X_vdj_all', 'X_vdj_expanded')
wrote `.obsp['connectivities']` & `['distances']` from graph[0]
stashed GEX matrices in `.uns['dandelion']` ('gex_connectivities', 'gex_distances')
stashed VDJ matrices in `.uns['dandelion']` under 'vdj_connectivities_*' keys
added `.uns['clone_id']` clone-level mapping (0:00:00)
[4]:
AnnData object with n_obs × n_vars = 25063 × 1309
obs: 'sample_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'gmm_pct_count_clusters_keep', 'scrublet_score', 'is_doublet', 'filter_rna', 'has_contig', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'leiden', 'clone_id', 'clone_id_rank'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'chain_status_colors', 'hvg', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'sample_id_colors', 'umap', 'dandelion', 'gex_neighbors', 'clone_id'
obsm: 'X_pca', 'X_umap', 'X_vdj'
varm: 'PCs'
obsp: 'connectivities', 'distances'
Note
If a column with the same name between Dandelion.metadata and AnnData.obs already exists, tl.transfer will not overwrite the column in the AnnData object. This can be toggled to overwrite all with overwrite=True or overwrite=["column_name1", "column_name2"] if only some columns are to be overwritten.
You can see that AnnData object now contains a couple more columns in the .obs slot, corresponding to the metadata that is returned after tl.generate_network, and newly populated .obsm and .obsp slots. The original RNA connectivities and distances are now added into the .obsp slot as well.
Plotting in scanpy
pl.clone_network
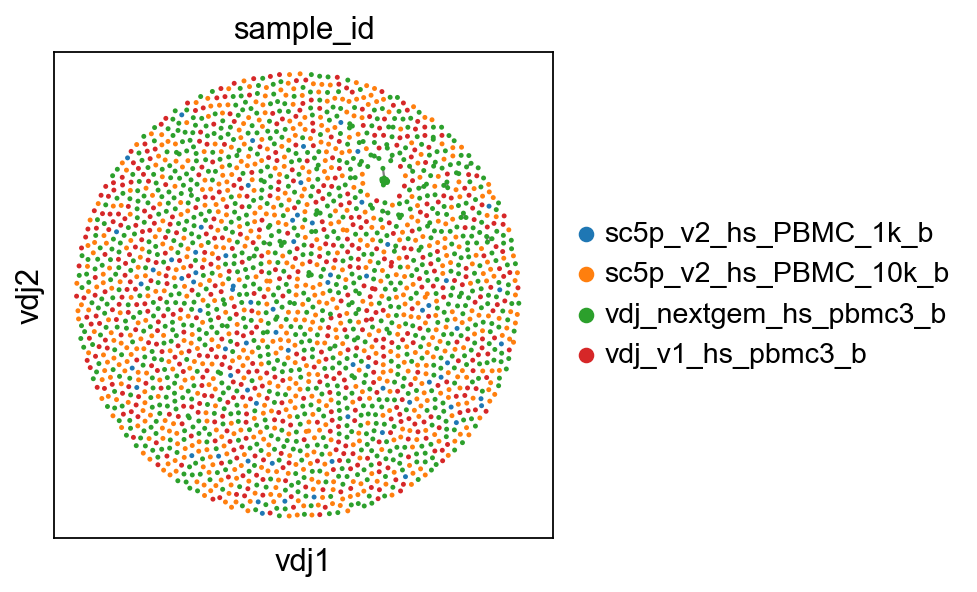
So now, basically we can plot in scanpy with their plotting modules. I’ve included a plotting function in dandelion, pl.clone_network, which is really just a wrapper of their pl.embedding module.
[5]:
sc.set_figure_params(figsize=[4, 4])
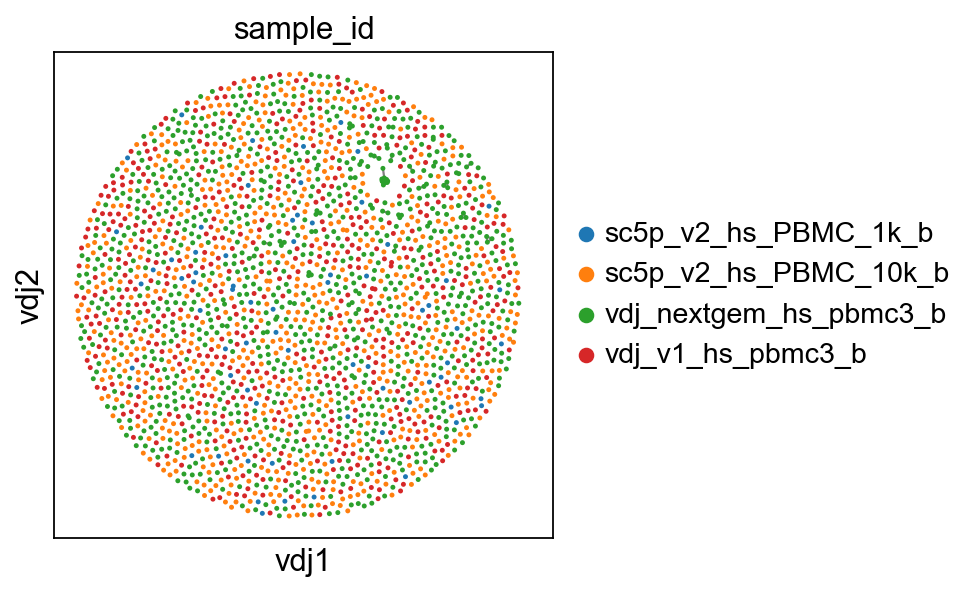
ddl.pl.clone_network(adata, color=["sample_id"], edges_width=1, size=20)
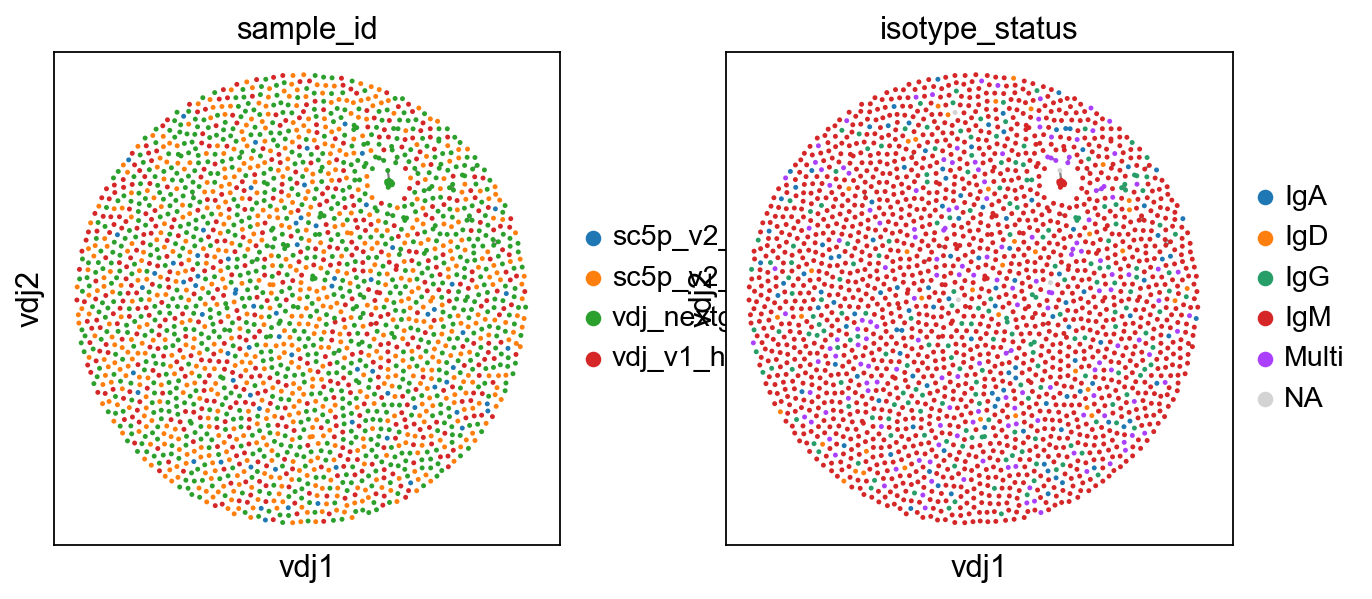
[6]:
ddl.pl.clone_network(
adata, color=["sample_id", "isotype_status"], edges_width=1, size=20
)
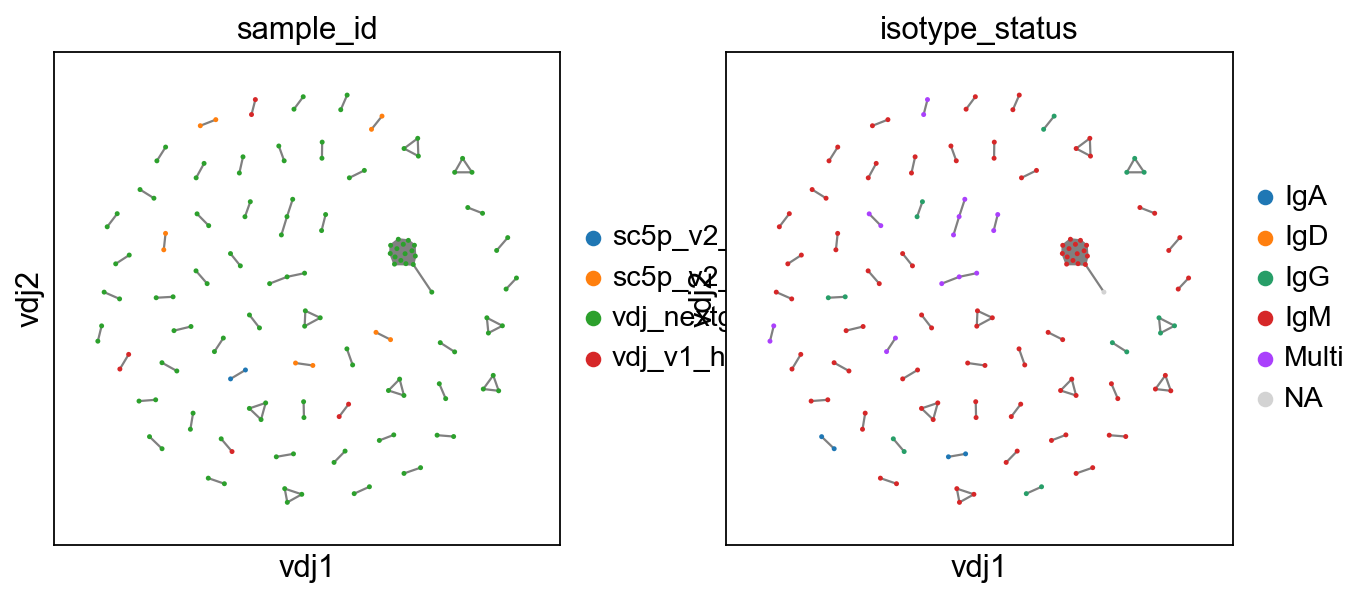
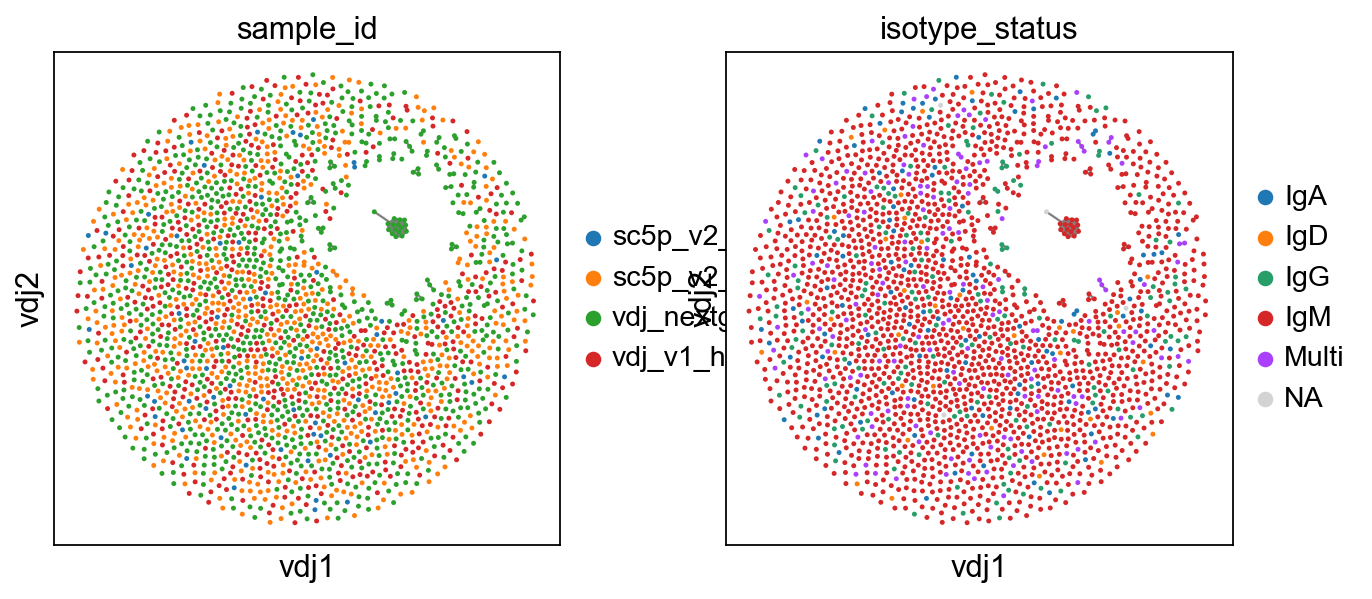
To show only expanded clones, we swap the view with a new function.
[7]:
ddl.tl.clone_view(adata, mode="expanded")
ddl.pl.clone_network(
adata, color=["sample_id", "isotype_status"], edges_width=1, size=20
)
[8]:
# swap back to "all"
ddl.tl.clone_view(adata, mode="all")
ddl.pl.clone_network(adata, color=["sample_id"], edges_width=1, size=20)
The mode can also be swap to rna to reset the neighborhood graph to the RNA-based one for any downstream analysis. If mode is set to None, then the user can specify the keys in .obsp and .obsm to set as active by providing connectivities_key, distances_key, and embedding_key.
Note
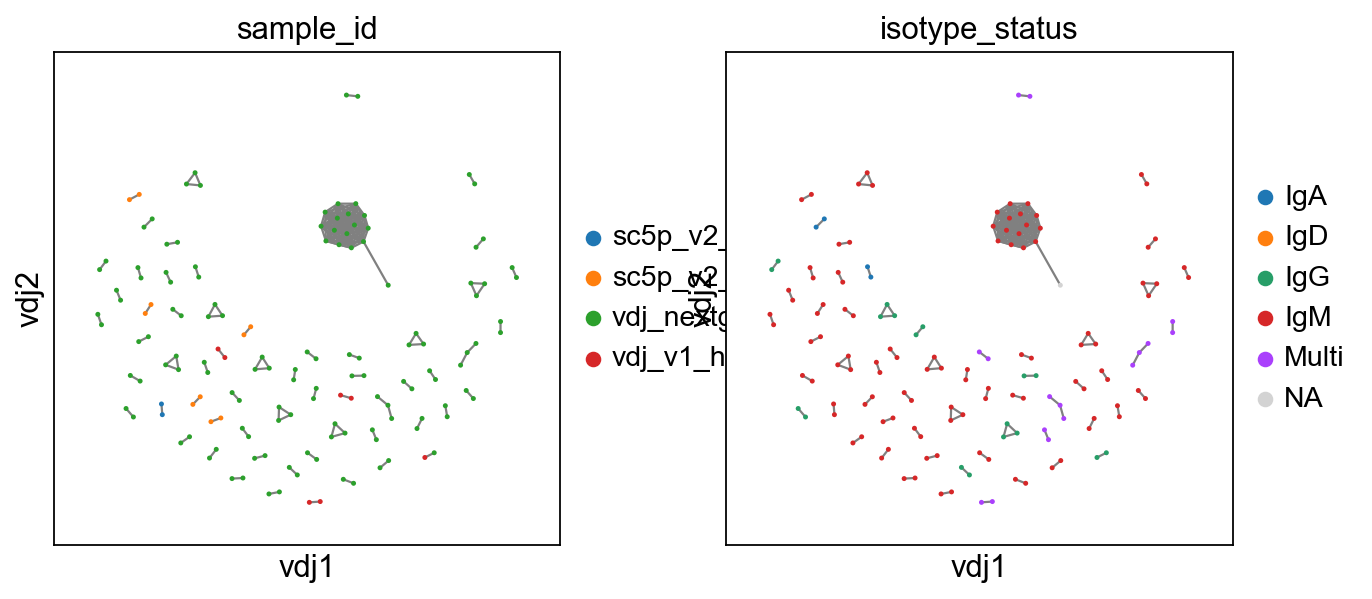
If you want a faster way to compute the layout for large graphs, you might want to try the mod_fr_bh layout method. Unlike the default mod_fr2 which uses a Numba-accelerated modified Fruchterman-Reingold algorithm, mod_fr_bh uses a Barnes-Hut approximation with O(N log N) complexity, making it more scalable for large datasets.
[9]:
# making a copy of both adata and vdj
vdj2 = vdj.copy()
adata2 = adata.copy()
# recompute layout with original method
ddl.tl.generate_network(vdj2, layout_method="mod_fr_bh")
ddl.tl.transfer(adata2, vdj2)
# visualise
ddl.pl.clone_network(
adata2, color=["sample_id", "isotype_status"], edges_width=1, size=20
)
# swap to expanded mode.
ddl.tl.clone_view(adata2, mode="expanded")
# show where clones/clonotypes have more than 1 cell
ddl.pl.clone_network(
adata2, color=["sample_id", "isotype_status"], edges_width=1, size=20
)
Generating network layout from pre-computed network
Computing network layout (Barnes-Hut CPU)
Computing expanded network layout (Barnes-Hut CPU)
finished.
Updated Dandelion object
: 'layout', graph layout
(0:00:00)
Transferring network
finished: updated `.obs` with `.metadata`
wrote active layout to `.obsm['X_vdj']`; stashed all views in `.uns['dandelion']` ('X_vdj_all', 'X_vdj_expanded')
wrote `.obsp['connectivities']` & `['distances']` from graph[0]
stashed GEX matrices in `.uns['dandelion']` ('gex_connectivities', 'gex_distances')
stashed VDJ matrices in `.uns['dandelion']` under 'vdj_connectivities_*' keys
added `.uns['clone_id']` clone-level mapping (0:00:00)
OMP: Info #276: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
tl.extract_edge_weights
We provide an edge weight extractor tool tl.extract_edge_weights to retrieve the edge weights that can be used to specify the edge widths according to weight/distance.
[10]:
# To illustrate this, first recompute the graph by specifying a minimum size
vdjx = vdj.copy()
adatax = adata.copy()
ddl.tl.generate_network(
vdjx, min_size=3
) # second graph will only contain clones/clonotypes with >= 3 cells
ddl.tl.transfer(adatax, vdjx)
edgeweights = [
1 / (e + 1) for e in ddl.tl.extract_edge_weights(vdjx)
] # invert and add 1 to each edge weight (e) so that distance of 0 becomes the thickest edge.
# therefore, the thicker the line, the shorter the edit distance.
ddl.tl.clone_view(adatax, mode="expanded")
ddl.pl.clone_network(
adatax,
color=["isotype_status"],
legend_fontoutline=3,
edges_width=edgeweights,
size=50,
)
Generating network
Setting up data: 4891it [00:00, 7354.27it/s]
Calculating distance matrix with distance_mode = 'clone'
100%|██████████| 2521/2521 [00:00<00:00, 15468.89it/s]
Distances calculated in 0.17 seconds
Sorting into clusters : 100%|██████████| 2521/2521 [00:00<00:00, 5999.69it/s]
Calculating minimum spanning tree : 100%|██████████| 64/64 [00:00<00:00, 1313.90it/s]
Generating edge list : 100%|██████████| 64/64 [00:00<00:00, 4225.07it/s]
Computing overlap : 100%|██████████| 2521/2521 [00:00<00:00, 2867.65it/s]
Adjust overlap : 100%|██████████| 154/154 [00:00<00:00, 4625.73it/s]
Linking edges : 100%|██████████| 2257/2257 [00:01<00:00, 1717.94it/s]
Computing network layout
Computing expanded network layout
finished.
Updated Dandelion object
: 'layout', graph layout
(0:00:18)
Transferring network
finished: updated `.obs` with `.metadata`
wrote active layout to `.obsm['X_vdj']`; stashed all views in `.uns['dandelion']` ('X_vdj_all', 'X_vdj_expanded')
wrote `.obsp['connectivities']` & `['distances']` from graph[0]
stashed GEX matrices in `.uns['dandelion']` ('gex_connectivities', 'gex_distances')
stashed VDJ matrices in `.uns['dandelion']` under 'vdj_connectivities_*' keys
added `.uns['clone_id']` clone-level mapping (0:00:00)
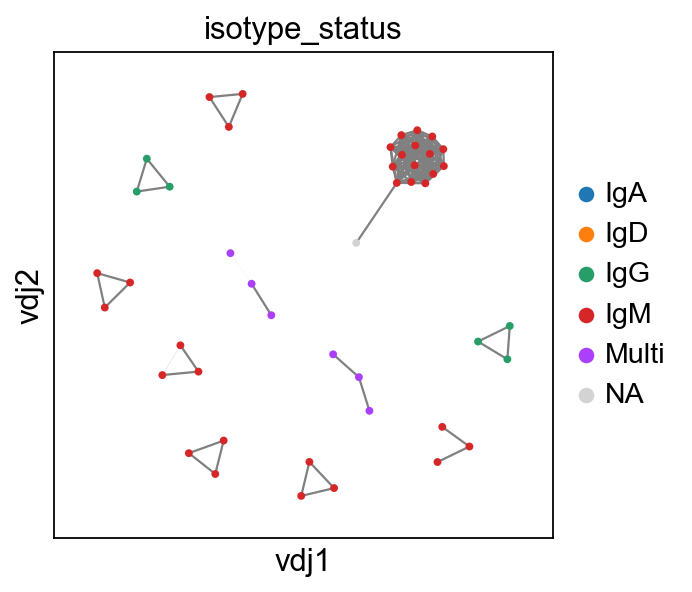
None here means there is no isotype information i.e. no c_call. If No_contig appears, it means there’s no V(D)J information.
You can interact with pl.clone_network just as how you interact with the rest of the scatterplot modules in scanpy.
[11]:
sc.set_figure_params(figsize=[4, 4.5])
ddl.pl.clone_network(
adata,
color=["locus_status", "chain_status"],
ncols=1,
legend_fontoutline=3,
edges_width=1,
size=20,
)
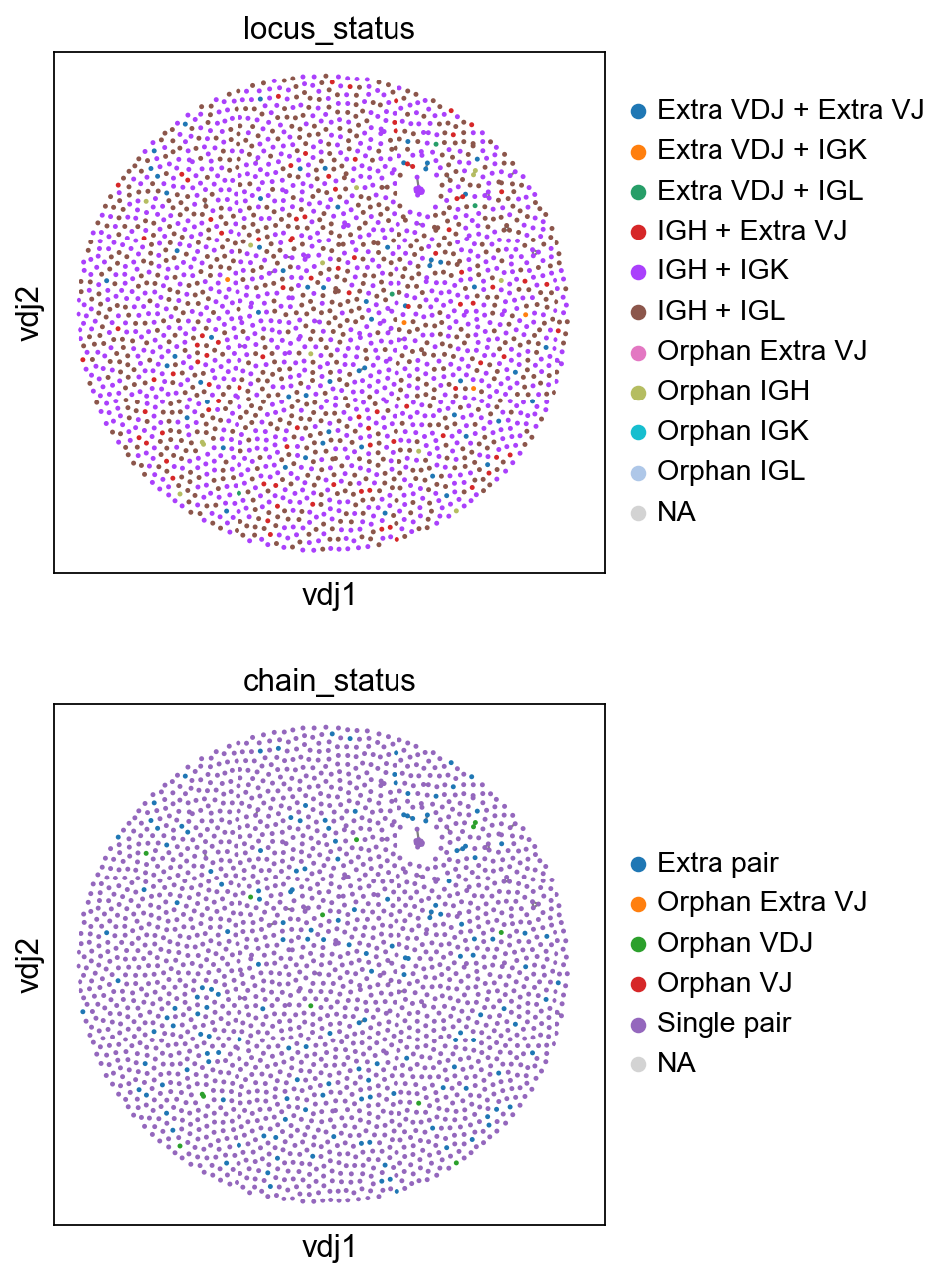
you should be able to save the adata object and interact with it as per normal.
[12]:
adata.write("adata.h5ad", compression="gzip")
Calculating size of clones
ddl.tl.clone_size
Sometimes it’s useful to evaluate the size of the clone. Here ddl.tl.clone_size does a simple calculation to enable that. From version 1.0.0 onwards, this function has been refactored and now also returning as proportion and can handle groups. There are new outputs e.g. clone size bins for rare, small, medium etc. similar to scRepertoire!
[13]:
ddl.tl.clone_size(vdj)
vdj.metadata[["clone_id_size", "clone_id_size_prop", "clone_id_size_category"]]
[13]:
| clone_id_size | clone_id_size_prop | clone_id_size_category | |
|---|---|---|---|
| sc5p_v2_hs_PBMC_10k_b_AAACCTGTCCGTTGTC | 1 | 0.000428 | Small |
| sc5p_v2_hs_PBMC_10k_b_AAACCTGTCGAGAACG | 1 | 0.000428 | Small |
| sc5p_v2_hs_PBMC_10k_b_AAACCTGTCTTGAGAC | 1 | 0.000428 | Small |
| sc5p_v2_hs_PBMC_10k_b_AAACGGGAGCGACGTA | 1 | 0.000428 | Small |
| sc5p_v2_hs_PBMC_10k_b_AAACGGGCACTGTTAG | 1 | 0.000428 | Small |
| ... | ... | ... | ... |
| vdj_v1_hs_pbmc3_b_TTTCCTCAGCGCTTAT | 1 | 0.000428 | Small |
| vdj_v1_hs_pbmc3_b_TTTCCTCAGGGAAACA | 1 | 0.000428 | Small |
| vdj_v1_hs_pbmc3_b_TTTCCTCTCGACAGCC | 1 | 0.000428 | Small |
| vdj_v1_hs_pbmc3_b_TTTGCGCCATACCATG | 1 | 0.000428 | Small |
| vdj_v1_hs_pbmc3_b_TTTGGTTGTAGGCATG | 1 | 0.000428 | Small |
2339 rows × 3 columns
[14]:
vdj.metadata["clone_id_size_category"].value_counts()
[14]:
clone_id_size_category
Small 2293
Medium 46
Name: count, dtype: int64
You can also compute the clone size within groups by providing the group_by argument.
[15]:
ddl.tl.clone_size(vdj, group_by="isotype_status")
[16]:
import pandas as pd
pd.crosstab(
vdj.metadata["isotype_status"], vdj.metadata["clone_id_size_category"]
).apply(lambda r: r / r.sum() * 100, axis=1)
[16]:
| clone_id_size_category | Hyperexpanded | Large | Medium | Small |
|---|---|---|---|---|
| isotype_status | ||||
| 100.0 | 0.000000 | 0.000000 | 0.000000 | |
| IgA | 0.0 | 3.448276 | 96.551724 | 0.000000 |
| IgD | 0.0 | 100.000000 | 0.000000 | 0.000000 |
| IgG | 0.0 | 9.090909 | 90.909091 | 0.000000 |
| IgM | 0.0 | 0.000000 | 5.879121 | 94.120879 |
| Multi | 0.0 | 9.876543 | 90.123457 | 0.000000 |
[17]:
ddl.tl.transfer(adata, vdj)
Transferring network
finished: updated `.obs` with `.metadata`
wrote active layout to `.obsm['X_vdj']`; stashed all views in `.uns['dandelion']` ('X_vdj_all', 'X_vdj_expanded')
wrote `.obsp['connectivities']` & `['distances']` from graph[0]
stashed GEX matrices in `.uns['dandelion']` ('gex_connectivities', 'gex_distances')
stashed VDJ matrices in `.uns['dandelion']` under 'vdj_connectivities_*' keys
added `.uns['clone_id']` clone-level mapping (0:00:00)
[18]:
sc.set_figure_params(figsize=[5, 4.5])
ddl.pl.clone_network(
adata,
color=["clone_id_size"],
legend_fontoutline=3,
edges_width=1,
size=20,
color_map="viridis",
)
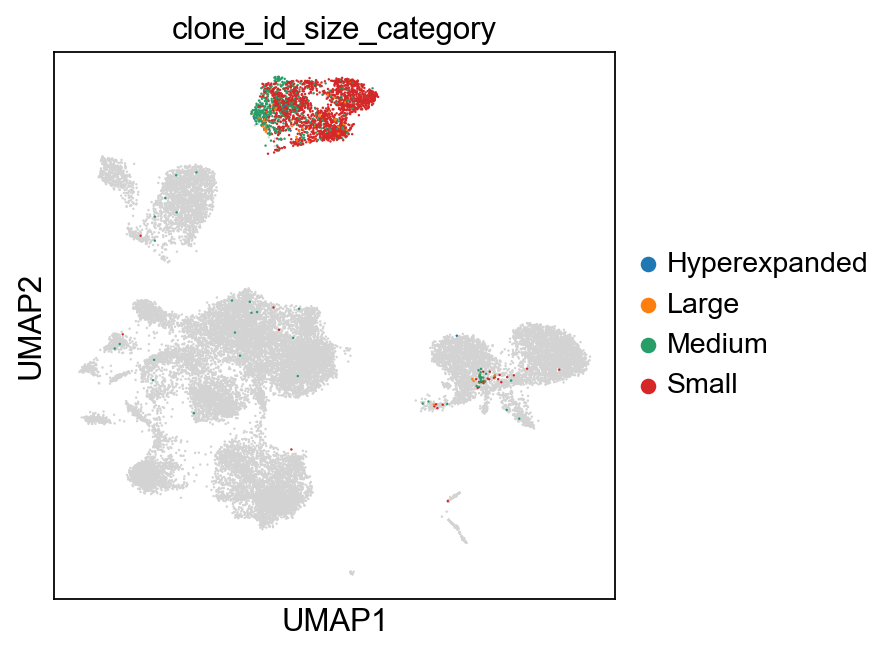
sc.pl.umap(adata, color=["clone_id_size"], color_map="viridis")
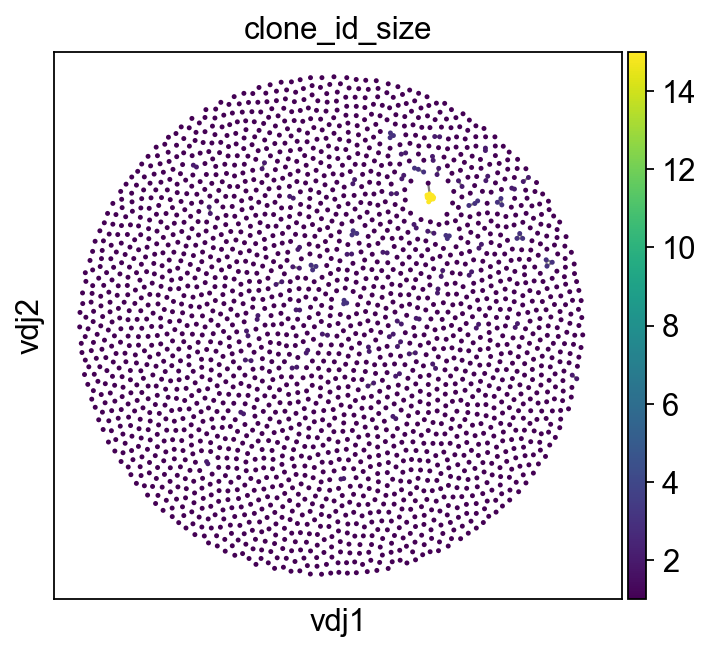
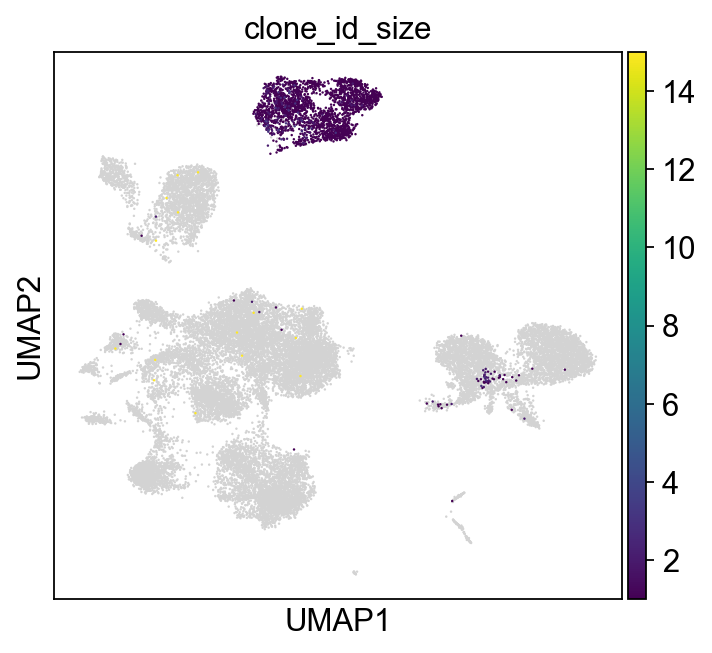
[19]:
sc.set_figure_params(figsize=[4.5, 4.5])
ddl.pl.clone_network(
adata,
color=["clone_id_size_category"],
legend_fontoutline=3,
edges_width=1,
size=20,
groups=["Small", "Medium", "Large", "Hyperexpanded"],
na_in_legend=False,
)
sc.pl.umap(
adata,
color=["clone_id_size_category"],
groups=["Small", "Medium", "Large", "Hyperexpanded"],
na_in_legend=False,
)
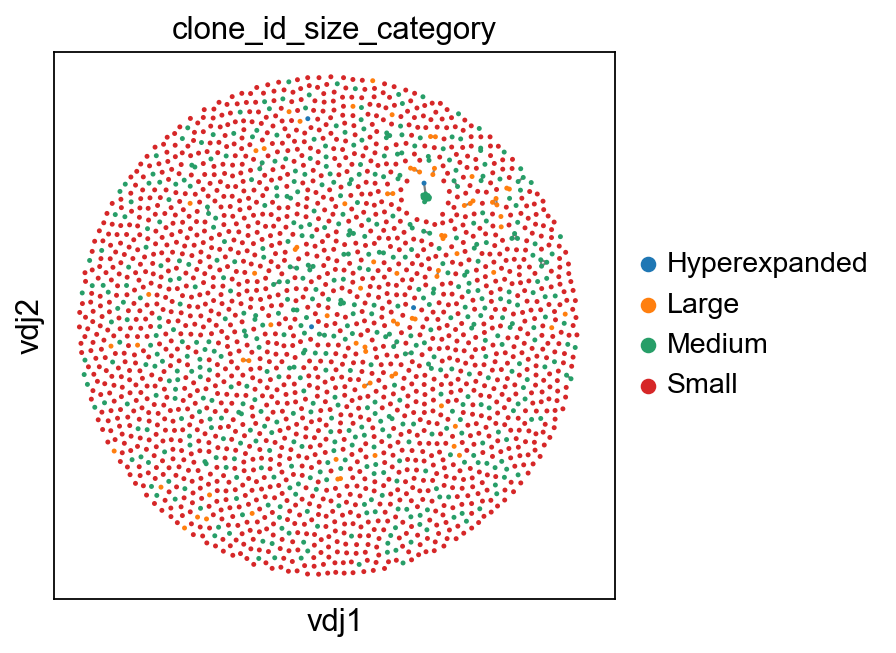
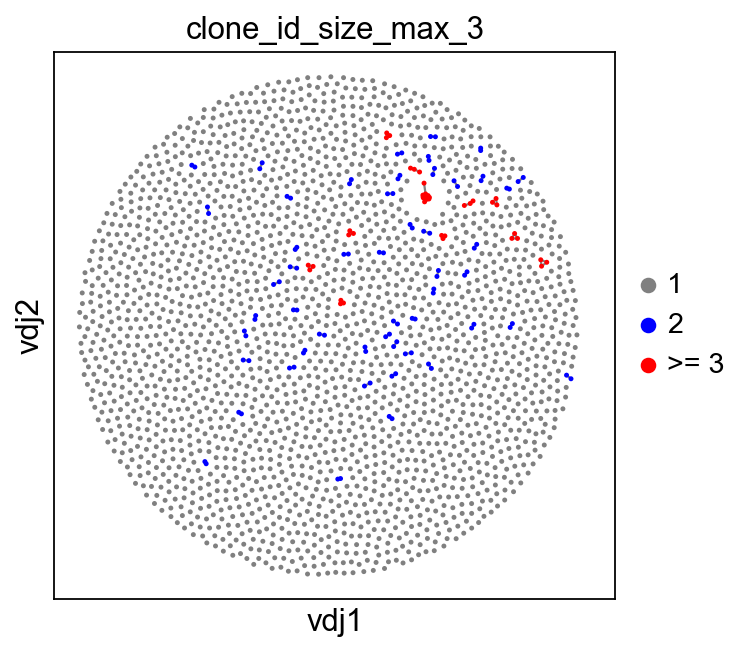
You can also specify max_size to clip off the calculation at a fixed value.
[20]:
ddl.tl.clone_size(vdj, max_size=3)
ddl.tl.transfer(adata, vdj)
Transferring network
finished: updated `.obs` with `.metadata`
wrote active layout to `.obsm['X_vdj']`; stashed all views in `.uns['dandelion']` ('X_vdj_all', 'X_vdj_expanded')
wrote `.obsp['connectivities']` & `['distances']` from graph[0]
stashed GEX matrices in `.uns['dandelion']` ('gex_connectivities', 'gex_distances')
stashed VDJ matrices in `.uns['dandelion']` under 'vdj_connectivities_*' keys
added `.uns['clone_id']` clone-level mapping (0:00:00)
[21]:
sc.set_figure_params(figsize=[4.5, 4.5])
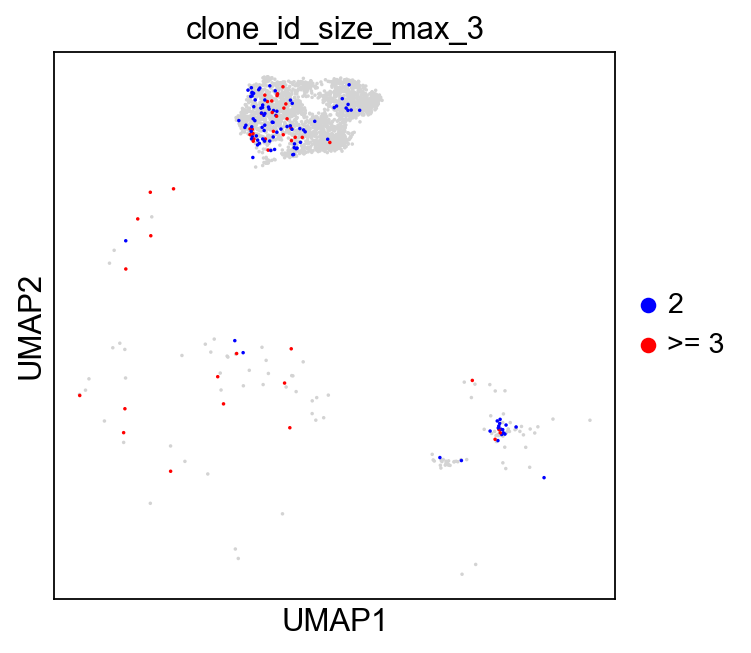
ddl.pl.clone_network(
adata,
color=["clone_id_size_max_3"],
ncols=2,
legend_fontoutline=3,
edges_width=1,
palette=["grey", "blue", "red"],
size=20,
na_in_legend=False,
)
sc.pl.umap(
adata[adata.obs["has_contig"] == "True"],
color=["clone_id_size_max_3"],
groups=["2", ">= 3"],
size=10,
na_in_legend=False,
)
Additional plotting functions
ddl.pl.barplot
pl.barplot is a generic barplot function that will plot items in the metadata slot as a bar plot. This function will also interact with .obs slot if a scanpy object is used in place of Dandelion object. However, if your scanpy object holds a lot of non-B cells, then the plotting will be just be saturated with nan values.
Let’s first slice the Dandelion object to only include cells with at most one heavy chain.
[22]:
vdj_subset = vdj[
vdj.metadata["chain_status"].isin(["Single pair", "Orphan VDJ"])
]
vdj_subset
[22]:
Dandelion class object with n_obs = 2177 and n_contigs = 4981
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'rearrangement_status', 'v_call_functionality', 'd_call_functionality', 'j_call_functionality', 'ambiguous', 'extra', 'clone_id'
metadata: 'clone_id', 'clone_id_rank', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'clone_id_size', 'clone_id_size_prop', 'clone_id_size_category', 'clone_id_size_max_3'
layout: layout for 2177 vertices, layout for 130 vertices
graph: networkx graph of 2177 vertices, networkx graph of 130 vertices
distances: distance matrix of shape (2177, 2177)
[23]:
import matplotlib as mpl
import matplotlib.pyplot as plt
mpl.rcParams.update(mpl.rcParamsDefault)
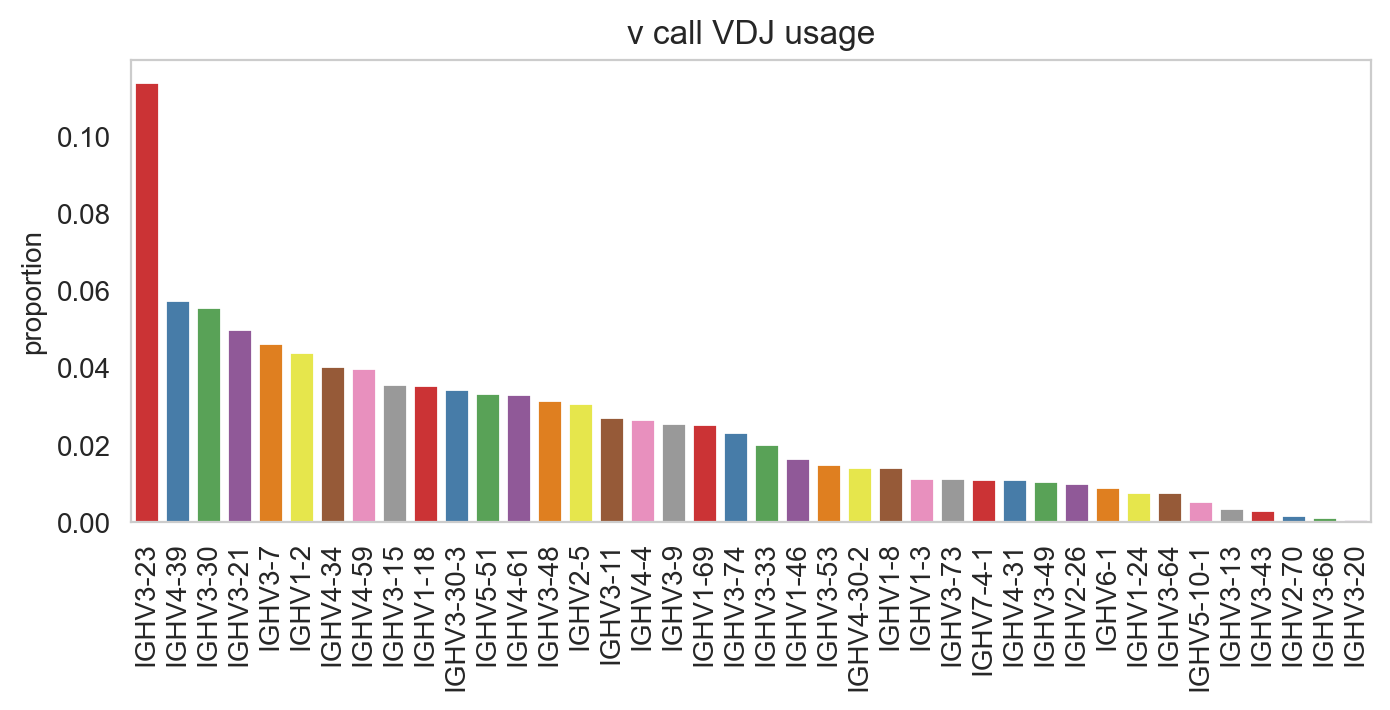
ddl.pl.barplot(
vdj_subset,
color="v_call_VDJ",
)
plt.show()
/Users/uqztuong/Documents/GitHub/dandelion/src/dandelion/base/plotting/_plotting.py:150: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
You can prevent it from sorting by specifying sort_descending = None. Colours can be changed with palette option.
[24]:
ddl.pl.barplot(
vdj_subset,
color="v_call_VDJ",
sort_descending=None,
palette="tab20",
)
plt.show()
/Users/uqztuong/Documents/GitHub/dandelion/src/dandelion/base/plotting/_plotting.py:150: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
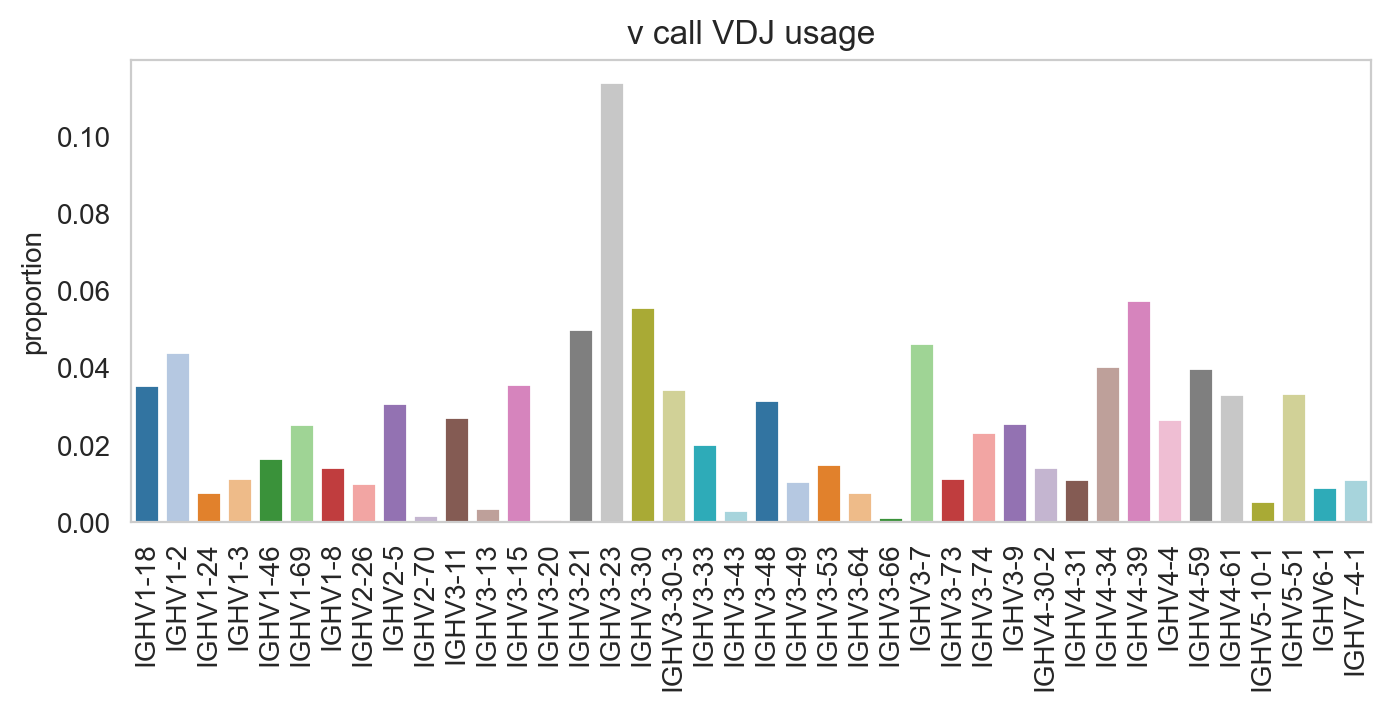
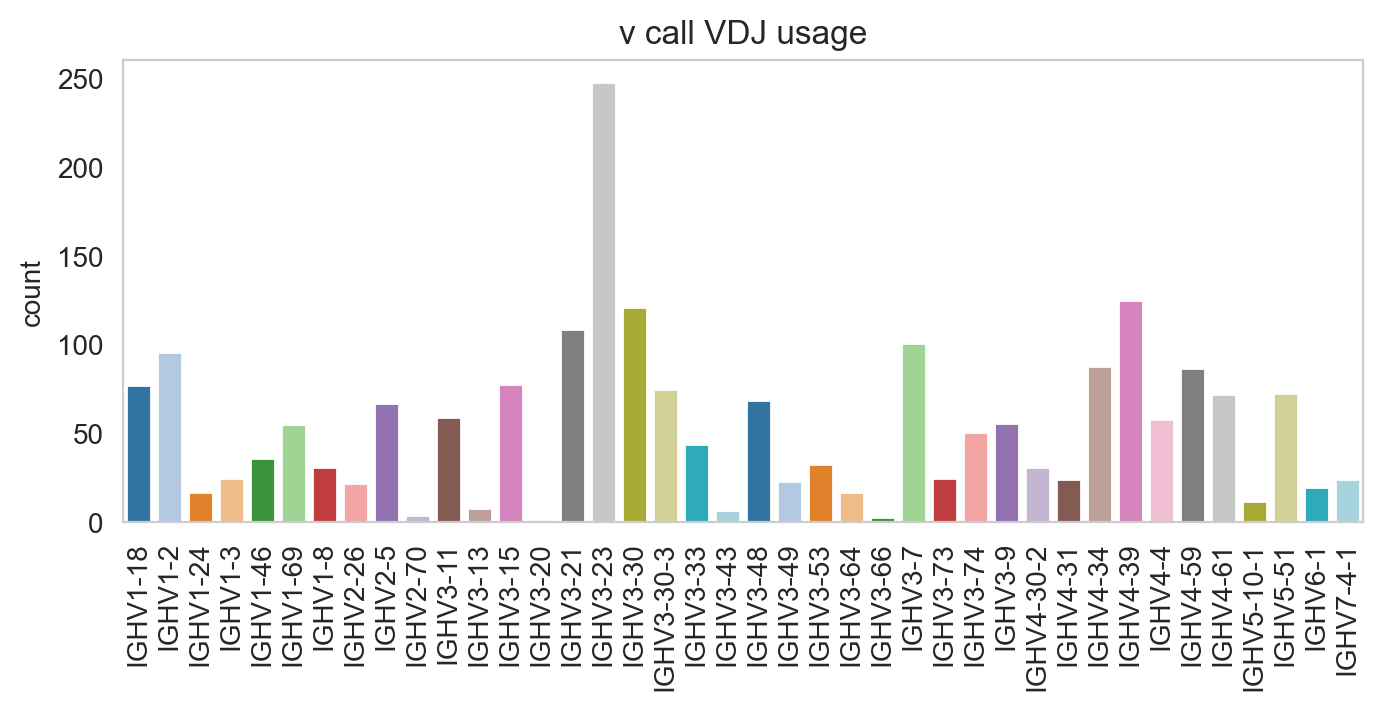
Specifying normalize = False will change the y-axis to counts.
[25]:
ddl.pl.barplot(
vdj_subset,
color="v_call_VDJ",
normalize=False,
sort_descending=None,
palette="tab20",
)
plt.show()
/Users/uqztuong/Documents/GitHub/dandelion/src/dandelion/base/plotting/_plotting.py:150: FutureWarning:
Passing `palette` without assigning `hue` is deprecated and will be removed in v0.14.0. Assign the `x` variable to `hue` and set `legend=False` for the same effect.
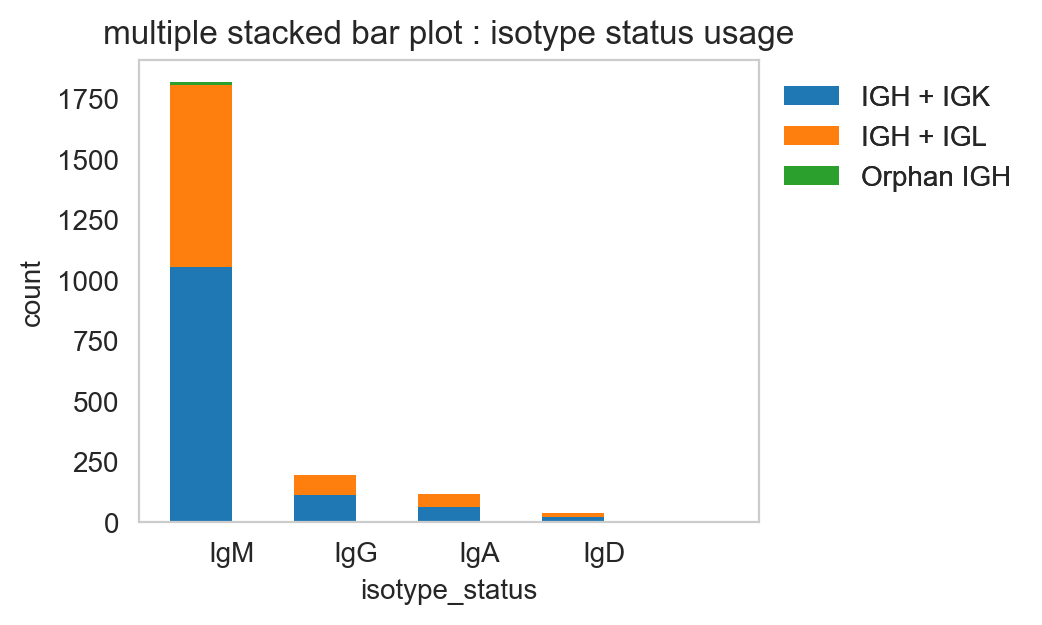
pl.stackedbarplot
pl.stackedbarplot is similar to above but can split between specified groups. Some examples below:
[26]:
ddl.pl.stackedbarplot(
vdj_subset,
color="isotype_status",
group_by="locus_status",
xtick_rotation=0,
figsize=(4, 3),
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
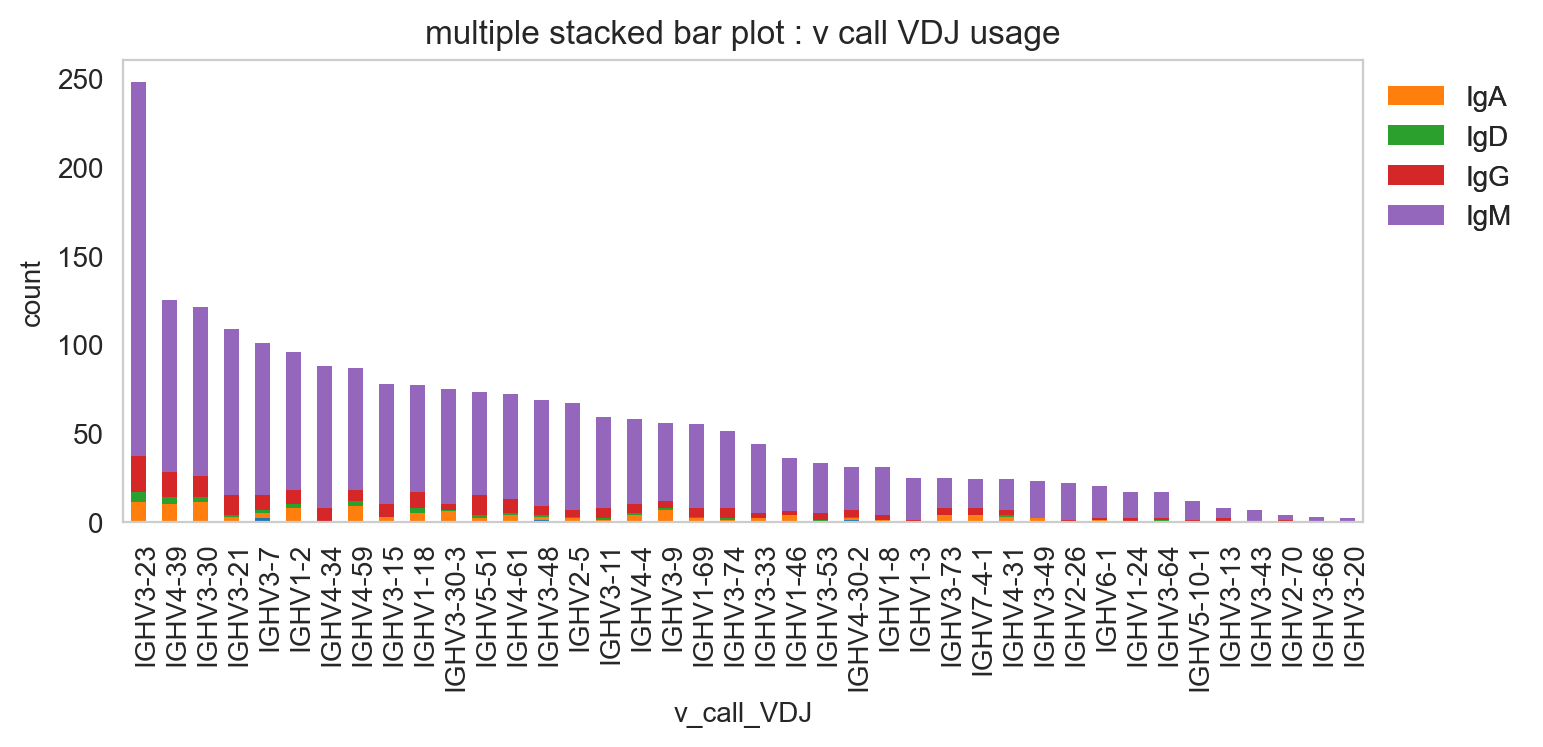
[27]:
ddl.pl.stackedbarplot(
vdj_subset,
color="v_call_VDJ",
group_by="isotype_status",
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
[28]:
ddl.pl.stackedbarplot(
vdj_subset,
color="v_call_VDJ",
group_by="isotype_status",
normalize=True,
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
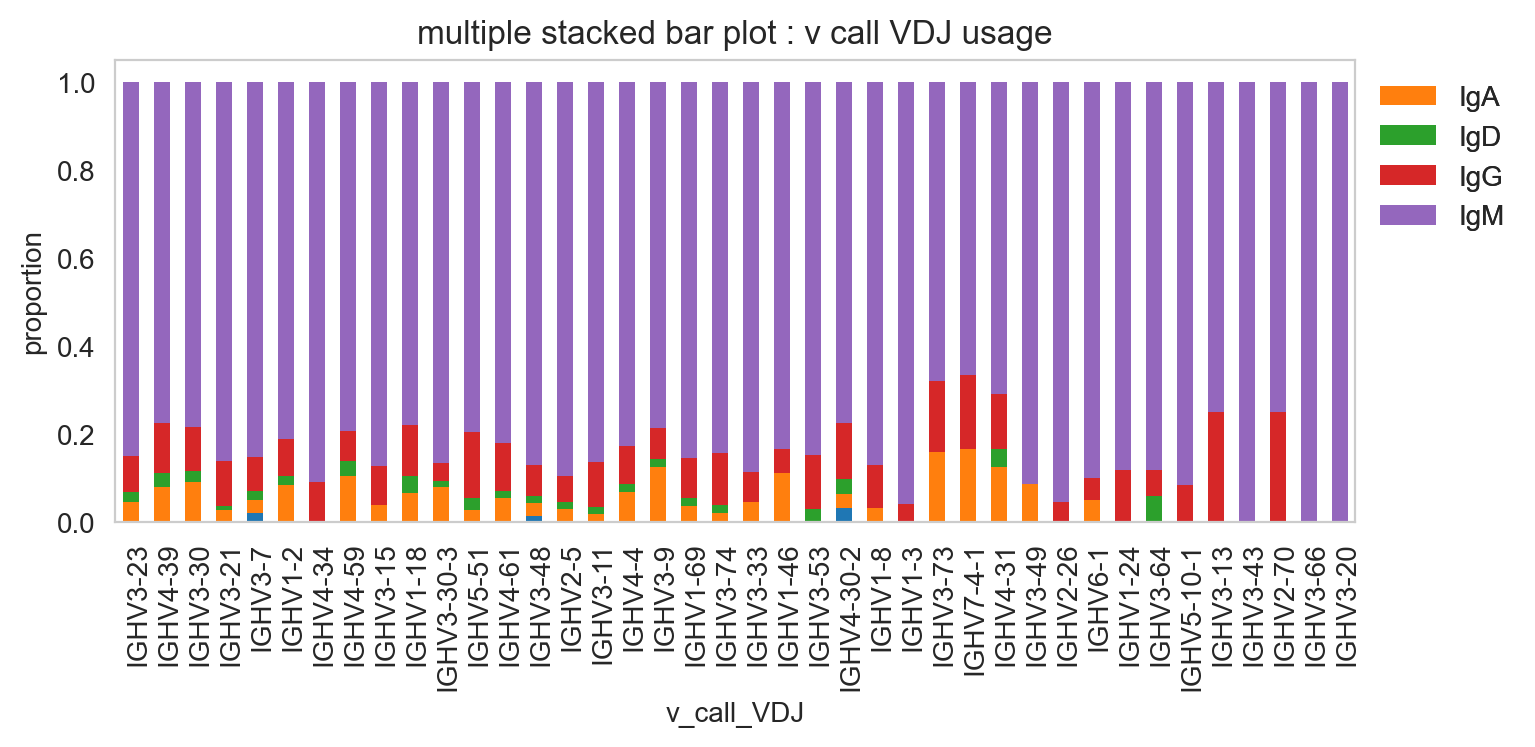
[29]:
ddl.pl.stackedbarplot(
vdj_subset,
color="v_call_VDJ",
group_by="chain_status",
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
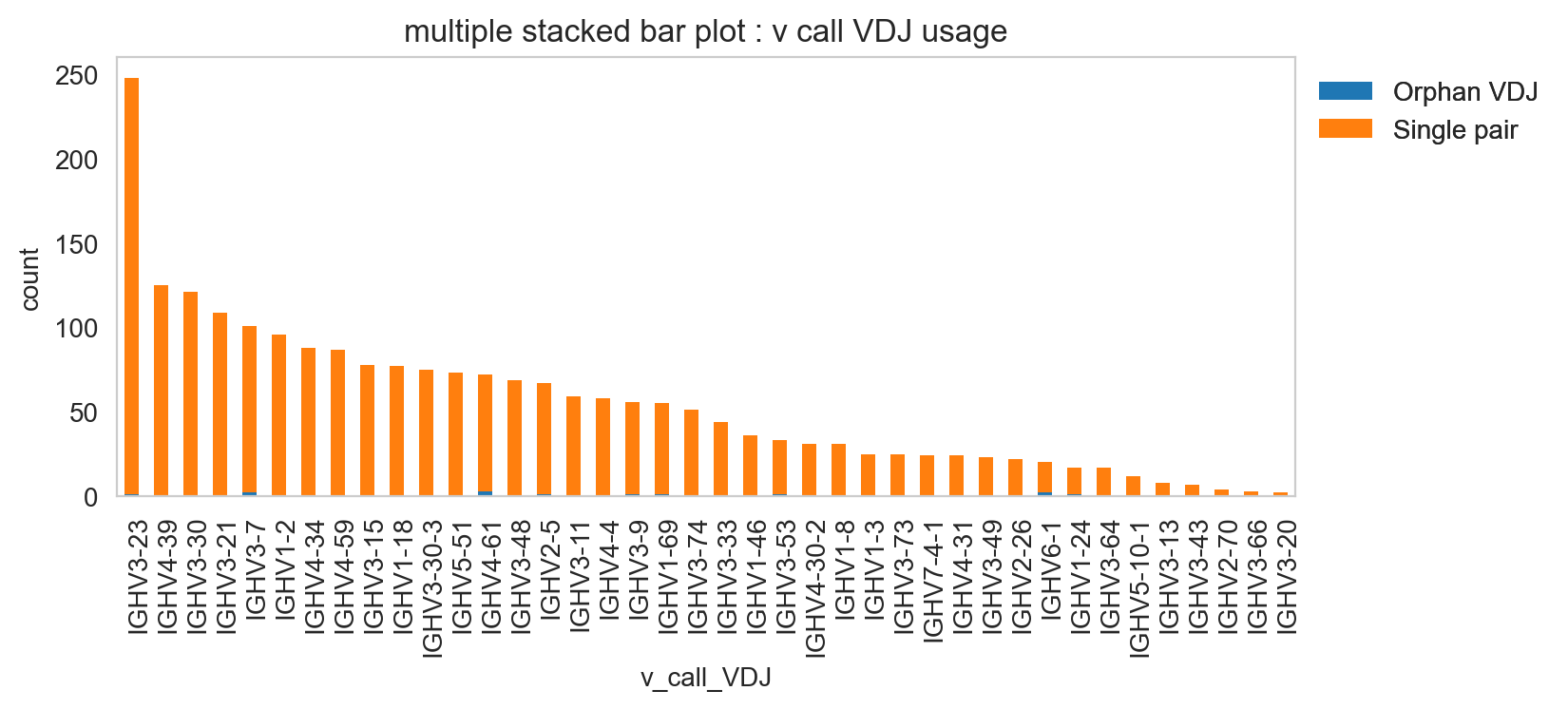
It’s obviously more useful if you don’t have too many groups, but you could try and plot everything and jiggle the legend options and color.
[30]:
ddl.pl.stackedbarplot(
vdj_subset,
color="v_call_VDJ",
group_by="sample_id",
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
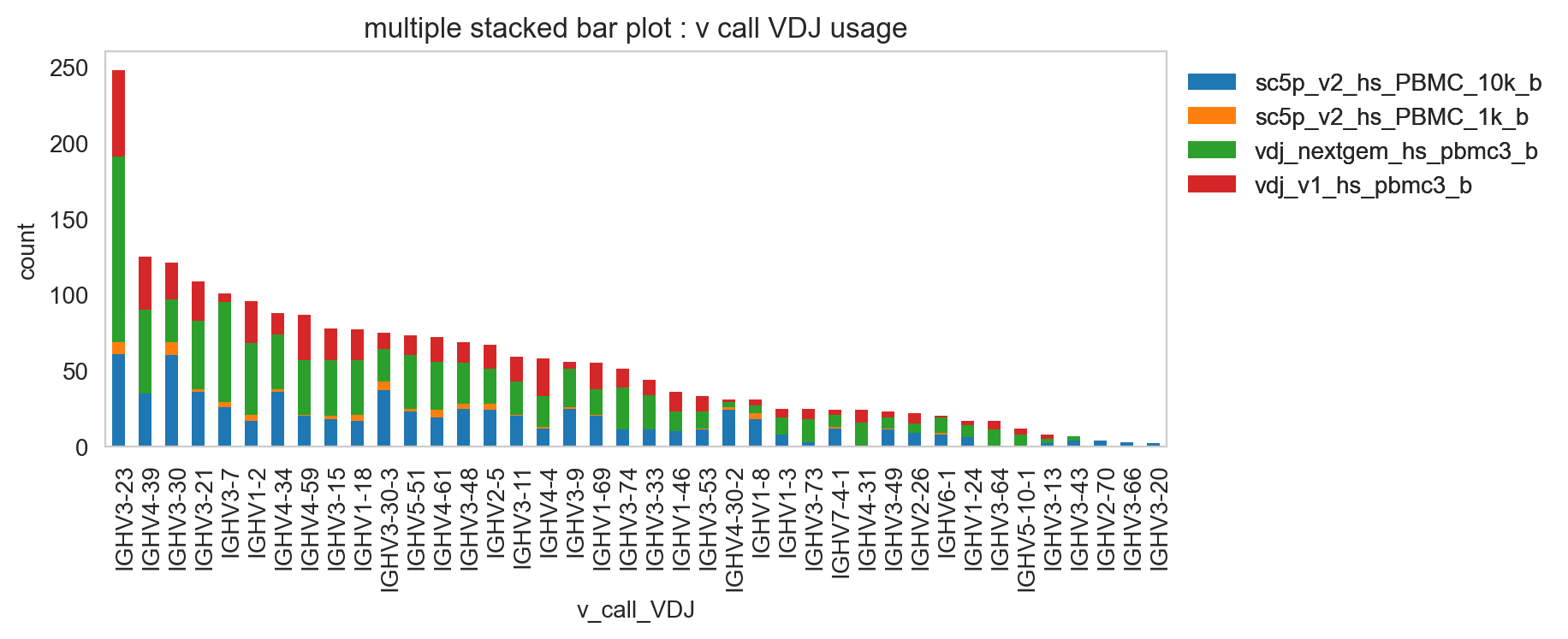
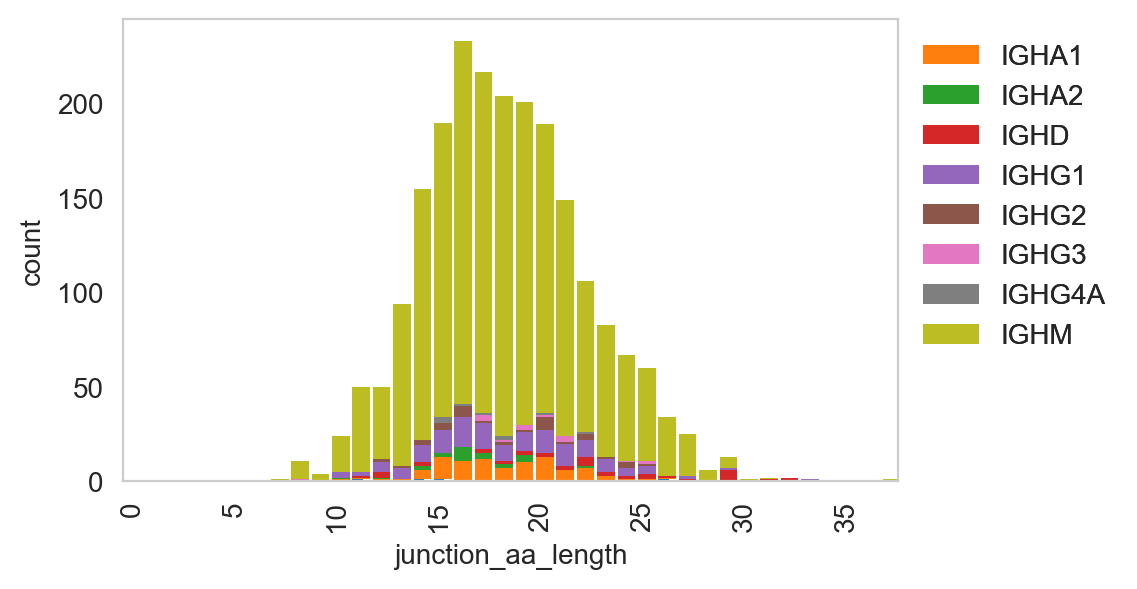
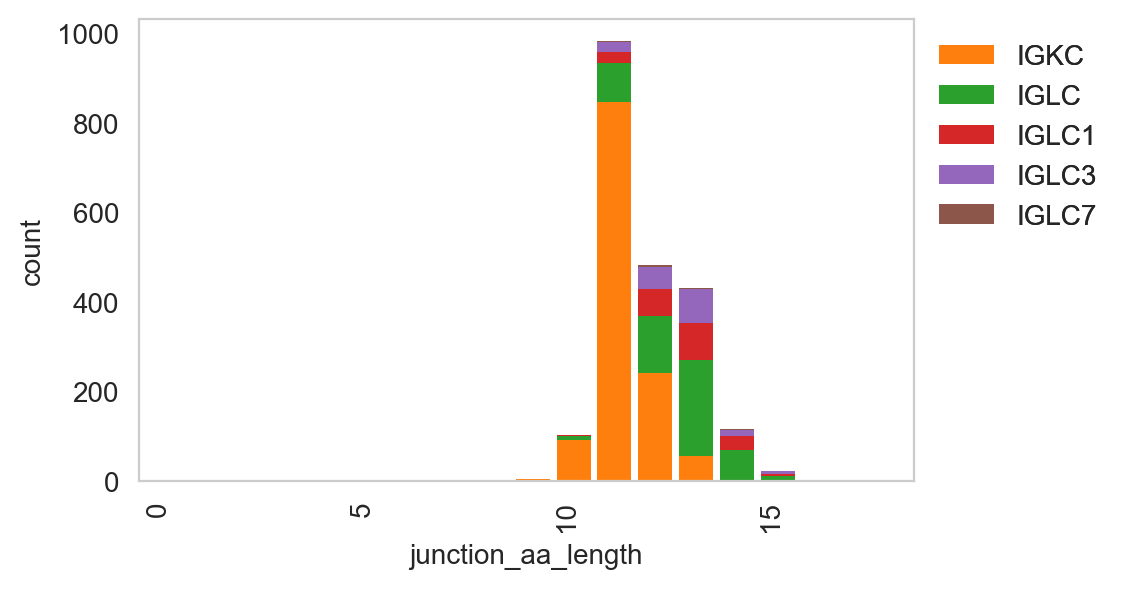
ddl.pl.spectratype
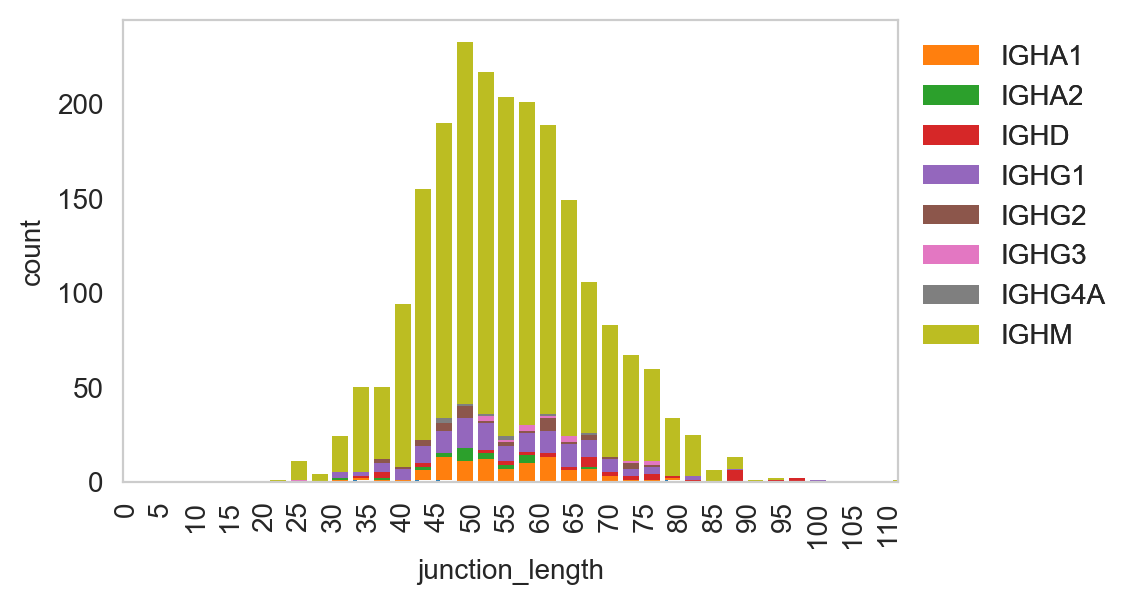
Spectratype plots contain info displaying CDR3 length distribution for specified groups. For this function, the current method only works for dandelion objects as it requires access to the contig-indexed .data slot.
[31]:
ddl.pl.spectratype(
vdj_subset,
color="junction_length",
group_by="c_call",
locus="IGH",
width=2.3,
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
[32]:
ddl.pl.spectratype(
vdj_subset,
color="junction_aa_length",
group_by="c_call",
locus="IGH",
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
[33]:
ddl.pl.spectratype(
vdj_subset,
color="junction_aa_length",
group_by="c_call",
locus=["IGK", "IGL"],
)
plt.legend(bbox_to_anchor=(1, 1), loc="upper left", frameon=False)
plt.show()
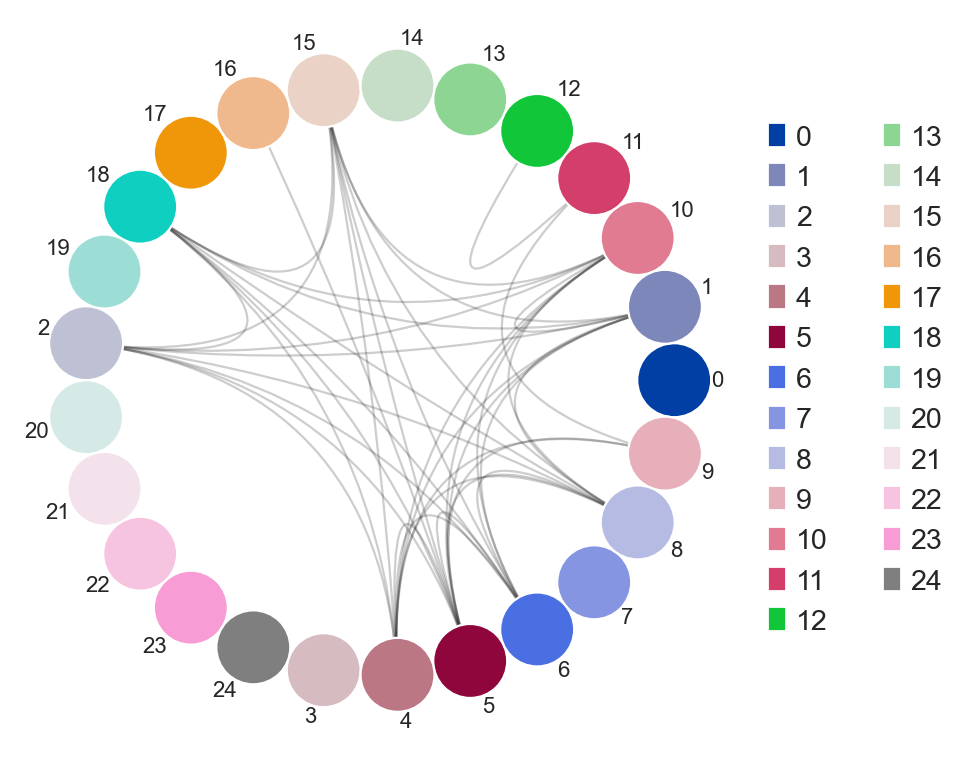
ddl.pl.clone_overlap
There is now a circos-style clone overlap function where it looks for whather different samples share a clone. If they do, an arc/connection will be drawn between them.
[34]:
ddl.tl.clone_overlap(adata, group_by="leiden")
Calculating clone overlap
finished: Updated AnnData:
'uns', clone overlap table (0:00:00)
[35]:
sc.set_figure_params(figsize=[6, 6])
ddl.pl.clone_overlap(adata, group_by="leiden")
plt.show()
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.12/site-packages/nxviz/annotate.py:68: FutureWarning: DataFrameGroupBy.apply operated on the grouping columns. This behavior is deprecated, and in a future version of pandas the grouping columns will be excluded from the operation. Either pass `include_groups=False` to exclude the groupings or explicitly select the grouping columns after groupby to silence this warning.
Other use cases for this would be, for example, to plot nodes as individual samples and the colors as group classifications of the samples. As long as this information is found in the .obs column in the AnnData, or even Dandelion.metadata, this will work.
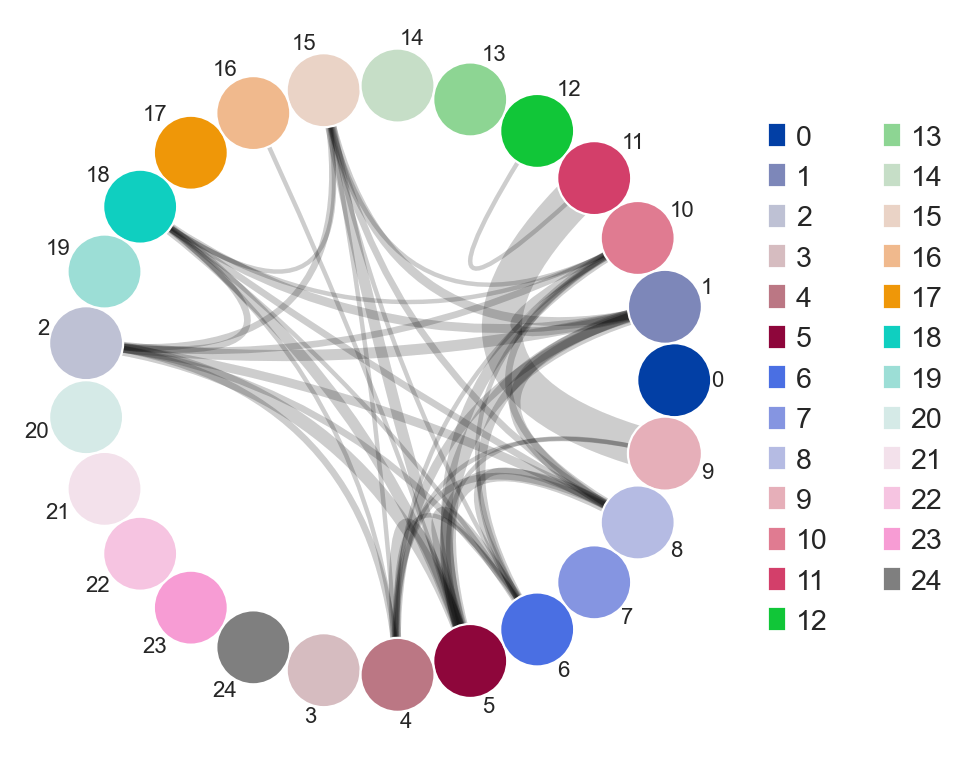
You an also specify weighted_overlap = True and the thickness of the edges will reflect the number of cells found to overlap between the nodes/samples.
[36]:
ddl.tl.clone_overlap(adata, group_by="leiden", weighted_overlap=True)
ddl.pl.clone_overlap(adata, group_by="leiden", weighted_overlap=True)
plt.show()
Calculating clone overlap
finished: Updated AnnData:
'uns', clone overlap table (0:00:00)
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.12/site-packages/nxviz/annotate.py:68: FutureWarning: DataFrameGroupBy.apply operated on the grouping columns. This behavior is deprecated, and in a future version of pandas the grouping columns will be excluded from the operation. Either pass `include_groups=False` to exclude the groupings or explicitly select the grouping columns after groupby to silence this warning.
You can also visualise this as a heatmap by specifying as_heatmap = True.
[37]:
import seaborn as sns
sns.set(font_scale=0.8)
ddl.pl.clone_overlap(
adata,
group_by="leiden",
weighted_overlap=True,
as_heatmap=True,
# seaborn clustermap kwargs
cmap="Blues",
annot=True,
figsize=(8, 8),
annot_kws={"size": 10},
fmt="g",
)
plt.show()

tl.vj_usage_pca
You can also compute the V/J gene usage in your various groups of interest. This function will return a new AnnData where instead of cells (obs) by gene (var), it will be group_by (obs) by V/J genes (var).
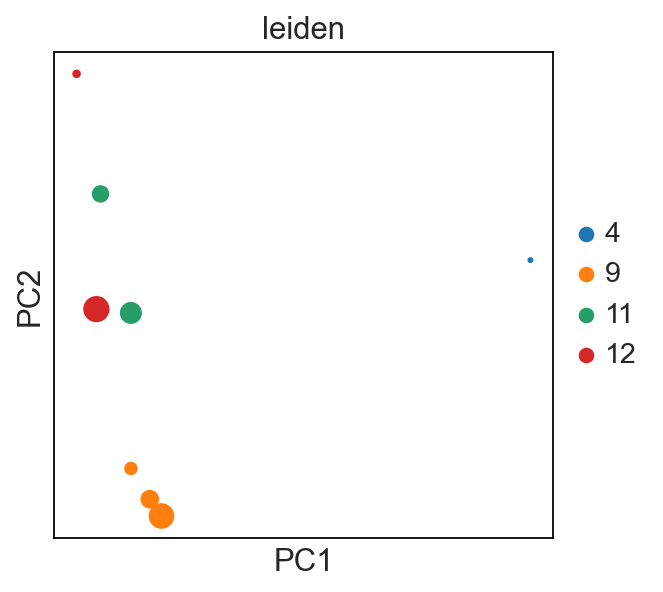
For example, I’m interested if the leiden clusters within each donor’s sample use V/J genes differently:
[38]:
# first make a concatenated group
adata.obs["sample_id_leiden"] = [
s + "_" + l for s, l in zip(adata.obs["sample_id"], adata.obs["leiden"])
]
new_adata = ddl.tl.vj_usage_pca(
adata,
group_by="sample_id_leiden",
mode="B", # because B cells, use abT and gdT for alpha-beta and gamma-delta T cells respectively
transfer_mapping=[
"sample_id",
"leiden",
], # this transfers the sample_id and leiden values separately. if not provided, only sample_id_leiden is transferred.
n_comps=3, # 3 because the example is small here. the default is set at 30
)
new_adata
Computing PCA for V/J gene usage
computing PCA
with n_comps=3
finished (0:00:00)
finished: Returned AnnData:
'obsm', X_pca for V/J gene usage (0:00:00)
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.12/site-packages/scanpy/preprocessing/_pca/__init__.py:226: FutureWarning: Argument `use_highly_variable` is deprecated, consider using the mask argument. Use_highly_variable=True can be called through mask_var="highly_variable". Use_highly_variable=False can be called through mask_var=None
[38]:
AnnData object with n_obs × n_vars = 8 × 124
obs: 'cell_type', 'cell_count', 'sample_id', 'leiden'
uns: 'pca'
obsm: 'X_pca'
varm: 'PCs'
[39]:
sc.set_figure_params()
sc.pl.pca(new_adata, color="leiden", size=new_adata.obs["cell_count"])
# each dot is a `sample_id_leiden`. Check the .obs
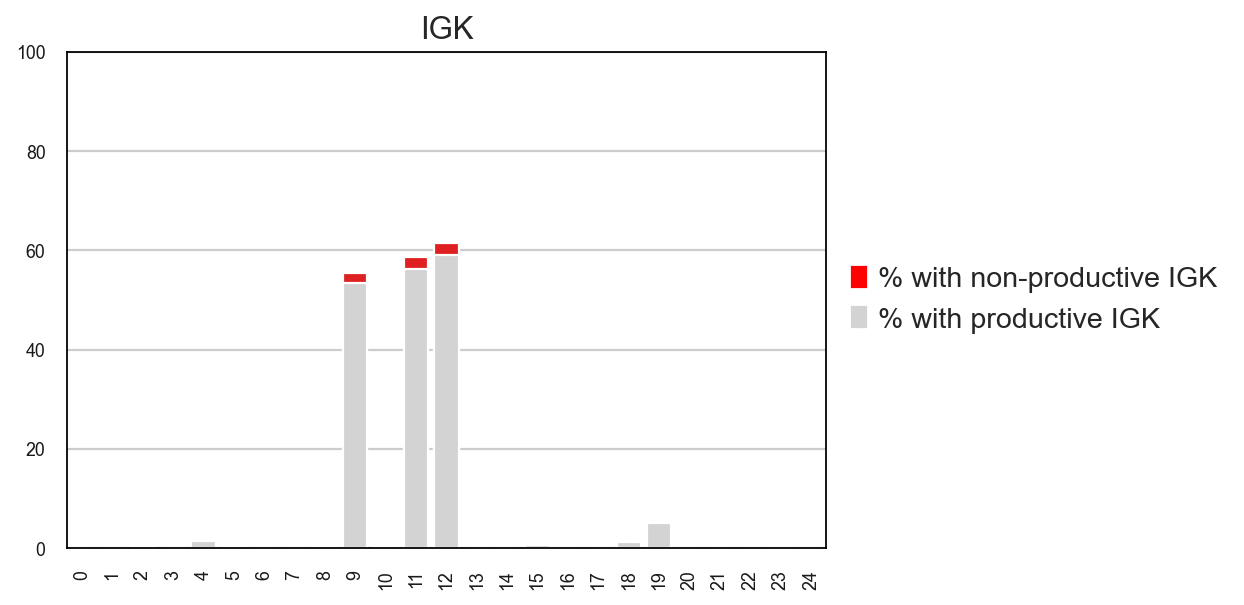
tl.productive_ratio/pl.productive_ratio
This new function lets you quantify what is the distribution of productive versus non-productive contigs at a cell-level. To do this, we need to re-check the Dandelion object so that non-productive columns are not removed.
[40]:
vdj2, adata2 = ddl.pp.check_contigs(vdj, adata, productive_only=False)
Filtering contigs
Preparing data: 5592it [00:00, 7890.10it/s]
Scanning for poor quality/ambiguous contigs: 100%|██████████| 2339/2339 [00:07<00:00, 310.48it/s]
Initializing Dandelion object
Transferring network
finished: updated `.obs` with `.metadata`
(0:00:00)
[41]:
ddl.tl.productive_ratio(adata2, vdj2, group_by="leiden", locus="IGK")
Tabulating productive ratio
finished: Updated AnnData:
'obs', 'IGK_productive'
'uns', 'productive_ratio'
(0:00:00)
[42]:
ddl.pl.productive_ratio(adata2, palette=["red", "lightgrey"])
plt.tight_layout()
# plt.savefig('plot.pdf')
plt.show()
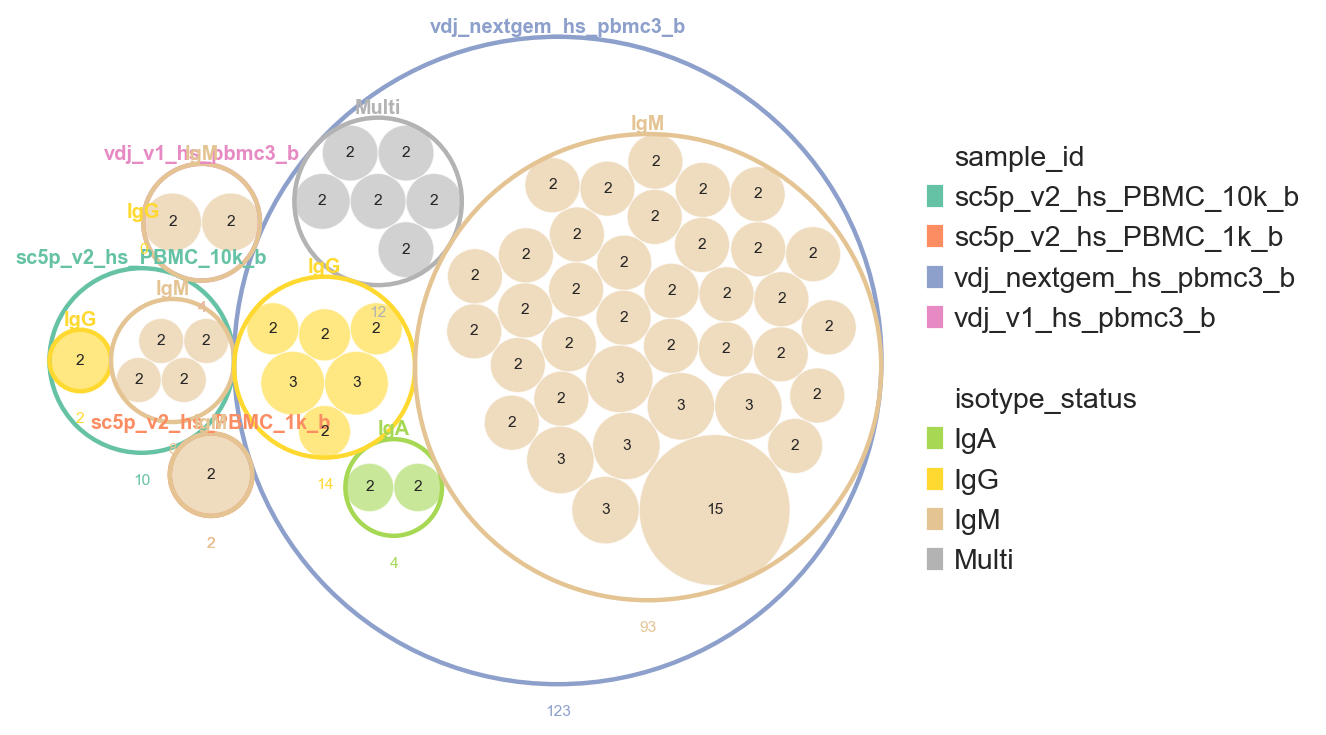
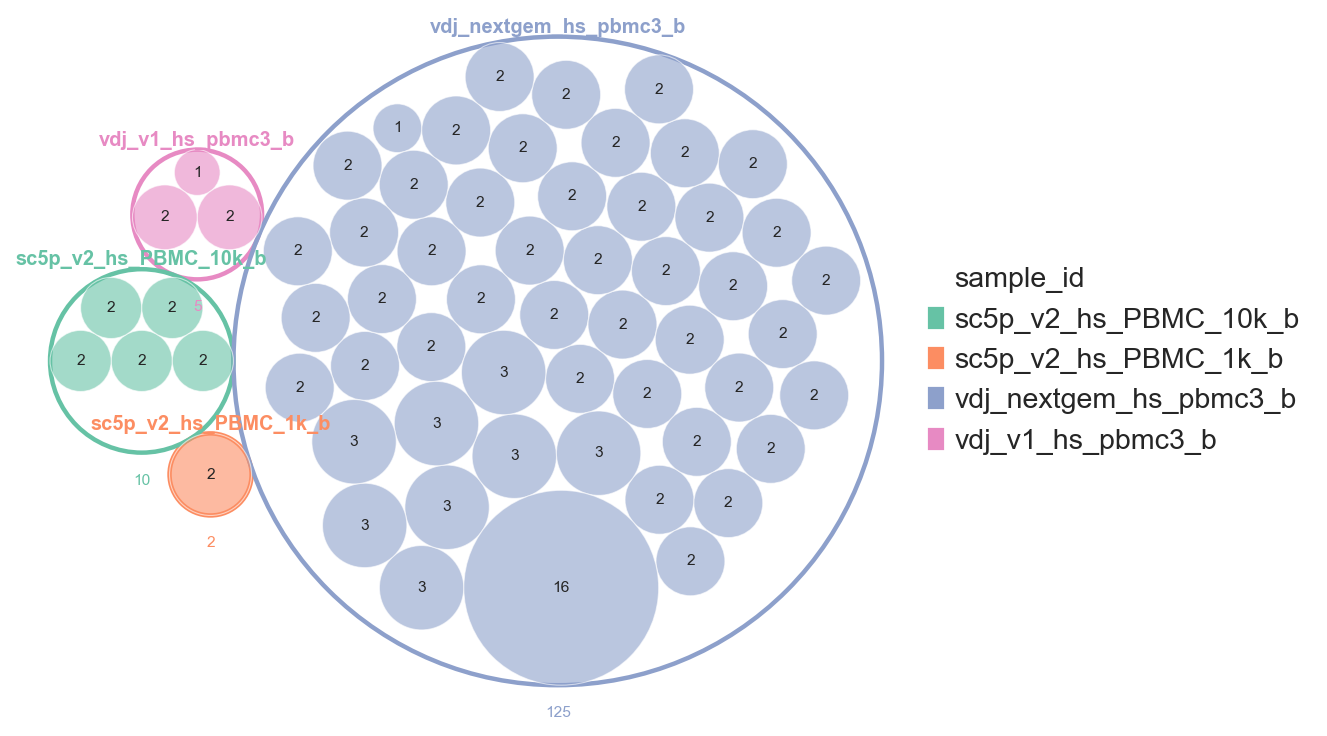
pl.clone_circlepackplot
pl.clone_circlepackplot is a bubble plot to visualise clone sizes within groups using circle packing. Each group (e.g. sample, celltype) is represented as an enclosing circle, with clones within that group shown as packed inner circles sized proportionally to clone size.
This function works with both AnnData and DandelionPolars objects. Singletons can be excluded by setting min_clone_size=2. We also enable show_count_labels=True to annotate each circle with its cell count.
[43]:
vdj.metadata["clone_id_size"].value_counts()
[43]:
clone_id_size
1 2193
2 100
3 30
16 16
Name: count, dtype: int64
[44]:
ddl.pl.clone_circlepackplot(
vdj,
group_by="sample_id",
min_clone_size=2,
show_count_labels=True,
figsize=(7, 7),
)
plt.show()
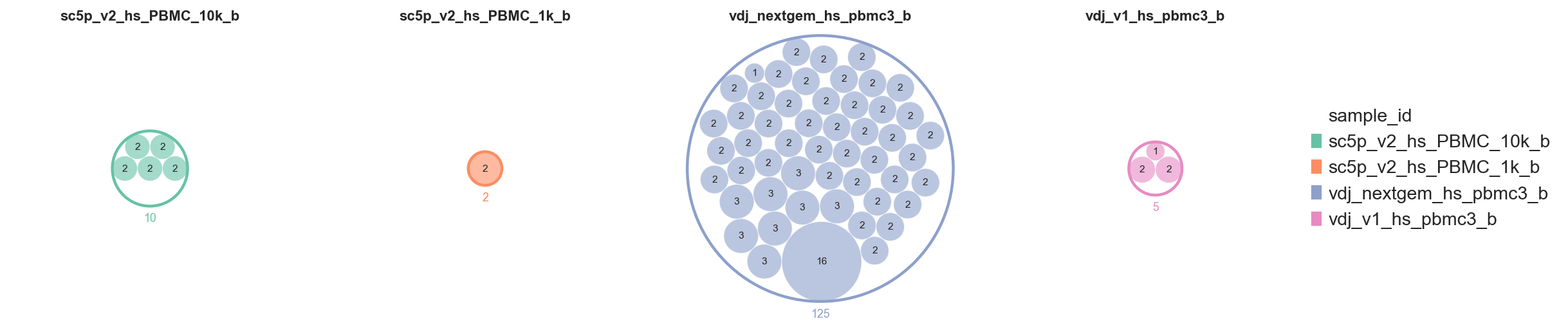
Setting as_subplots=True splits the figure into one panel per top-level group. scale_subplots=True (the default) scales enclosing circles proportionally to total cell count, so a clone of a given size always occupies the same area across panels.
[45]:
ddl.pl.clone_circlepackplot(
vdj,
group_by="sample_id",
min_clone_size=2,
as_subplots=True,
show_count_labels=True,
figsize=(4, 4),
)
plt.show()
Passing a list to group_by produces a multi-level nesting. The hierarchy follows the list order: the first element is the outermost ring, subsequent elements are nested rings, and clone circles sit at the innermost level. Each level is coloured independently using its own colour map.
[46]:
ddl.pl.clone_circlepackplot(
vdj,
group_by=["sample_id", "isotype_status"],
min_clone_size=2,
show_count_labels=True,
figsize=(7, 7),
)
plt.show()
min to max ratio is too low at 0.0 and it could cause algorithm stability issues. Try to remove insignificant data