V(D)J clustering
On the topic of finding clones/clonotypes, there are many ways used for clustering BCRs, almost all involving some measure based on sequence similarity. There are also a lot of very well established guidelines and criterias maintained by the BCR community. For example, immcantation uses a number of model-based methods [Gupta2015] to group clones based on the distribution of length-normalised junctional hamming distance while others use the whole BCR V(D)J sequence to define clones as shown in this paper [Bashford-Rogers2019].
Import modules
[22]:
import os
import dandelion as ddl
ddl.logging.print_header()
dandelion==0.5.5.dev16 pandas==2.2.3 numpy==2.1.3 matplotlib==3.10.1 networkx==3.4.2 scipy==1.15.2
[23]:
# change directory to somewhere more workable
os.chdir(os.path.expanduser("~/Downloads/dandelion_tutorial/"))
# I'm importing scanpy here to make use of its logging module.
import scanpy as sc
sc.settings.verbosity = 3
import warnings
warnings.filterwarnings("ignore")
sc.logging.print_header()
scanpy==1.10.3 anndata==0.11.3 umap==0.5.7 numpy==2.1.3 scipy==1.15.2 pandas==2.2.3 scikit-learn==1.6.1 statsmodels==0.14.4 igraph==0.11.8 pynndescent==0.5.13
Read in the previously saved files
I will work with the same example from the previous section since I have the filtered V(D)J data stored in a Dandelion class.
[24]:
vdj = ddl.read_h5ddl("dandelion_results.h5ddl")
vdj
[24]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status'
metadata: 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
Finding clones
The following is dandelion’s implementation of a rather conventional method to define clones, ddl.tl.find_clones.
Clone definition criterion
Clone definition is based on the following criterion:
Identical V- and J-gene usage in the VDJ chain (IGH/TRB/TRD).
Identical CDR3 junctional/CDR3 sequence length in the VDJ chain.
VDJ chain junctional/CDR3 sequences attains a minimum of % sequence similarity, based on hamming distance. The similarity cut-off is tunable (default is 85%; change to 100% if analyzing TCR data).
VJ chain (IGK/IGL/TRA/TRG) usage. If cells within clones use different VJ chains, the clone will be split following the same conditions for VDJ chains in (1-3) as above.
(Optional) Simplifying annotation calls.
This is an optional step to simplify the annotation calls. The vdj.simplify() will reduce the complexity of the gene names and strip the alleles. This impacts on how clones are defined (using simpler v/j calls) and also visualiation.
[25]:
# original
vdj.data[["v_call_genotyped", "j_call", "c_call"]]
[25]:
| v_call_genotyped | j_call | c_call | |
|---|---|---|---|
| sequence_id | |||
| sc5p_v2_hs_PBMC_10k_AAACCTGTCATATCGG_contig_1 | IGKV1-33*01,IGKV1D-33*01 | IGKJ4*01 | IGKC |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCCGTTGTC_contig_2 | IGHV1-69*01,IGHV1-69D*01 | IGHJ3*02 | IGHM |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCCGTTGTC_contig_1 | IGKV1-8*01 | IGKJ1*01 | IGKC |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCGAGAACG_contig_1 | IGLV5-45*02 | IGLJ3*02 | IGLC3 |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCGAGAACG_contig_2 | IGHV1-2*02 | IGHJ3*02 | IGHM |
| ... | ... | ... | ... |
| vdj_v1_hs_pbmc3_TTTCCTCTCGACAGCC_contig_1 | IGHV1-46*01 | IGHJ5*02 | IGHM |
| vdj_v1_hs_pbmc3_TTTGCGCCATACCATG_contig_2 | IGHV1-69*01,IGHV1-69D*01 | IGHJ6*02 | IGHM |
| vdj_v1_hs_pbmc3_TTTGCGCCATACCATG_contig_1 | IGLV1-47*01 | IGLJ3*02 | IGLC3 |
| vdj_v1_hs_pbmc3_TTTGGTTGTAGGCATG_contig_2 | IGLV2-11*01 | IGLJ2*01,IGLJ3*01,IGLJ3*02 | IGLC |
| vdj_v1_hs_pbmc3_TTTGGTTGTAGGCATG_contig_1 | IGHV3-23*01,IGHV3-23D*01 | IGHJ4*02 | IGHM |
7355 rows × 3 columns
[26]:
vdj.simplify()
vdj.data[["v_call_genotyped", "j_call", "c_call"]]
[26]:
| v_call_genotyped | j_call | c_call | |
|---|---|---|---|
| sequence_id | |||
| sc5p_v2_hs_PBMC_10k_AAACCTGTCATATCGG_contig_1 | IGKV1-33 | IGKJ4 | IGKC |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCCGTTGTC_contig_2 | IGHV1-69 | IGHJ3 | IGHM |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCCGTTGTC_contig_1 | IGKV1-8 | IGKJ1 | IGKC |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCGAGAACG_contig_1 | IGLV5-45 | IGLJ3 | IGLC3 |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCGAGAACG_contig_2 | IGHV1-2 | IGHJ3 | IGHM |
| ... | ... | ... | ... |
| vdj_v1_hs_pbmc3_TTTCCTCTCGACAGCC_contig_1 | IGHV1-46 | IGHJ5 | IGHM |
| vdj_v1_hs_pbmc3_TTTGCGCCATACCATG_contig_2 | IGHV1-69 | IGHJ6 | IGHM |
| vdj_v1_hs_pbmc3_TTTGCGCCATACCATG_contig_1 | IGLV1-47 | IGLJ3 | IGLC3 |
| vdj_v1_hs_pbmc3_TTTGGTTGTAGGCATG_contig_2 | IGLV2-11 | IGLJ2 | IGLC |
| vdj_v1_hs_pbmc3_TTTGGTTGTAGGCATG_contig_1 | IGHV3-23 | IGHJ4 | IGHM |
7355 rows × 3 columns
[27]:
# let's save this as a separate file
vdj.write_h5ddl("dandelion_results_simplified.h5ddl", compression="gzip")
Running ddl.tl.find_clones
The function will take a file path, a pandas DataFrame (for example if you’ve used pandas to read in the filtered file already), or a Dandelion class object. The default mode for calculation of junctional hamming distance is to use the CDR3 junction amino acid sequences, specified via the key option (None defaults to junction_aa). You can switch it to using CDR3 junction nucleotide sequences (key = 'junction'), or even the full V(D)J amino acid sequence
(key = 'sequence_alignment_aa'), as long as the column name exists in the .data slot.
Clustering TCR is possible with the same setup but requires changing of default parameters (covered in the TCR section).
[28]:
ddl.tl.find_clones(vdj)
vdj
Finding clonotypes
Finding clones based on B cell VDJ chains : 100%|██████████| 194/194 [00:00<00:00, 4210.54it/s]
Finding clones based on B cell VJ chains : 100%|██████████| 203/203 [00:00<00:00, 4665.80it/s]
Refining clone assignment based on VJ chain pairing : 100%|██████████| 2238/2238 [00:00<00:00, 551778.29it/s]
finished: Updated Dandelion object:
'data', contig-indexed AIRR table
'metadata', cell-indexed observations table
(0:00:00)
[28]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status', 'clone_id'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
This will return a new column with the column name 'clone_id' as per convention. If a file path is provided as input, it will also save the file automatically into the base directory of the file name. Otherwise, a Dandelion object will be returned.
Clonotype definition criterion
The clone_id follows an A_B_C_D_E_F format and largely reflects the conditions above where:
{A} indicates if the contigs use the same V and J genes in the VDJ chain.
{B} indicates if junctional/CDR3 sequences are equal in length in the VDJ chain.
{C} indicates if clones are split based on junctional/CDR3 hamming distance threshold (for VDJ chain).
{D} indicates if the contigs use the same V and J genes in the VJ chain.
{E} indicates if junctional/CDR3 sequences are equal in length in the VJ chain.
{F} indicates if clones are split based on junctional/CDR3 hamming distance threshold (for VJ chain).
Also, to prevent issues with clonotype ids matching between B cells and T cells, there will be a prefix added to the clone_id to reflect whether or not it’s a B, abT or gdT clone.
Also, to reduce ambiguity, the A_B_C segment will have the VDJ prefix and the D_E_F segment will have the VJ suffix.
Therefor, a complete B cell clonotype id will look something like:
B_VDJ_1_1_2_VJ_2_1_1
For an Orphan VDJ, it would be B_VDJ_1_1_2.
For an Orphan VJ, it would be B_VJ_2_1_1.
There is also an alternate column called clone_id_by_size which is a simple numerical version of the clone_id which corresponds to the size of the clonotype - 1 is the largest clonotype, 2 is the second largest, and so on.
[29]:
vdj.metadata
[29]:
| clone_id | clone_id_by_size | sample_id | locus_VDJ | locus_VJ | productive_VDJ | productive_VJ | v_call_genotyped_VDJ | d_call_VDJ | j_call_VDJ | ... | d_call_B_VDJ_main | j_call_B_VDJ_main | v_call_B_VJ_main | j_call_B_VJ_main | isotype | isotype_status | locus_status | chain_status | rearrangement_status_VDJ | rearrangement_status_VJ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sc5p_v2_hs_PBMC_10k_AAACCTGTCATATCGG | B_VJ_186_2_8 | 169 | sc5p_v2_hs_PBMC_10k | None | IGK | None | T | None | None | None | ... | None | None | IGKV1-33 | IGKJ4 | Orphan IGK | Orphan VJ | None | standard | ||
| sc5p_v2_hs_PBMC_10k_AAACCTGTCCGTTGTC | B_VDJ_129_4_2_VJ_142_2_3 | 1989 | sc5p_v2_hs_PBMC_10k | IGH | IGK | T | T | IGHV1-69 | IGHD3-22 | IGHJ3 | ... | IGHD3-22 | IGHJ3 | IGKV1-8 | IGKJ1 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCGAGAACG | B_VDJ_81_1_2_VJ_41_1_1 | 1603 | sc5p_v2_hs_PBMC_10k | IGH | IGL | T | T | IGHV1-2 | None | IGHJ3 | ... | None | IGHJ3 | IGLV5-45 | IGLJ3 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
| sc5p_v2_hs_PBMC_10k_AAACCTGTCTTGAGAC | B_VDJ_130_4_5_VJ_30_1_1 | 1604 | sc5p_v2_hs_PBMC_10k | IGH | IGK | T | T | IGHV5-51 | None | IGHJ3 | ... | None | IGHJ3 | IGKV1D-8 | IGKJ2 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| sc5p_v2_hs_PBMC_10k_AAACGGGAGCGACGTA | B_VDJ_46_2_1_VJ_101_2_7 | 1605 | sc5p_v2_hs_PBMC_10k | IGH | IGL | T | T | IGHV4-4 | IGHD6-13 | IGHJ3 | ... | IGHD6-13 | IGHJ3 | IGLV3-19 | IGLJ2 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| vdj_v1_hs_pbmc3_TTTCCTCAGCAATATG | B_VDJ_174_1_1_VJ_71_2_8 | 813 | vdj_v1_hs_pbmc3 | IGH | IGK | T | T | IGHV2-5 | IGHD5/OR15-5a | IGHJ4 | ... | IGHD5/OR15-5a | IGHJ4 | IGKV4-1 | IGKJ4 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| vdj_v1_hs_pbmc3_TTTCCTCAGCGCTTAT | B_VDJ_93_5_6_VJ_153_1_3 | 814 | vdj_v1_hs_pbmc3 | IGH | IGK | T | T | IGHV3-30 | IGHD4-17 | IGHJ6 | ... | IGHD4-17 | IGHJ6 | IGKV2-30 | IGKJ2 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| vdj_v1_hs_pbmc3_TTTCCTCAGGGAAACA | B_VDJ_83_1_1_VJ_83_4_13 | 815 | vdj_v1_hs_pbmc3 | IGH | IGK | T | T | IGHV4-61 | IGHD6-13 | IGHJ2 | ... | IGHD6-13 | IGHJ2 | IGKV1-39 | IGKJ1 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| vdj_v1_hs_pbmc3_TTTGCGCCATACCATG | B_VDJ_38_7_2_VJ_160_3_5 | 816 | vdj_v1_hs_pbmc3 | IGH | IGL | T | T | IGHV1-69 | IGHD2-15 | IGHJ6 | ... | IGHD2-15 | IGHJ6 | IGLV1-47 | IGLJ3 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
| vdj_v1_hs_pbmc3_TTTGGTTGTAGGCATG | B_VDJ_144_5_4_VJ_79_3_2 | 2389 | vdj_v1_hs_pbmc3 | IGH | IGL | T | T | IGHV3-23 | None | IGHJ4 | ... | None | IGHJ4 | IGLV2-11 | IGLJ2 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
2238 rows × 47 columns
Alternative : Running tl.define_clones
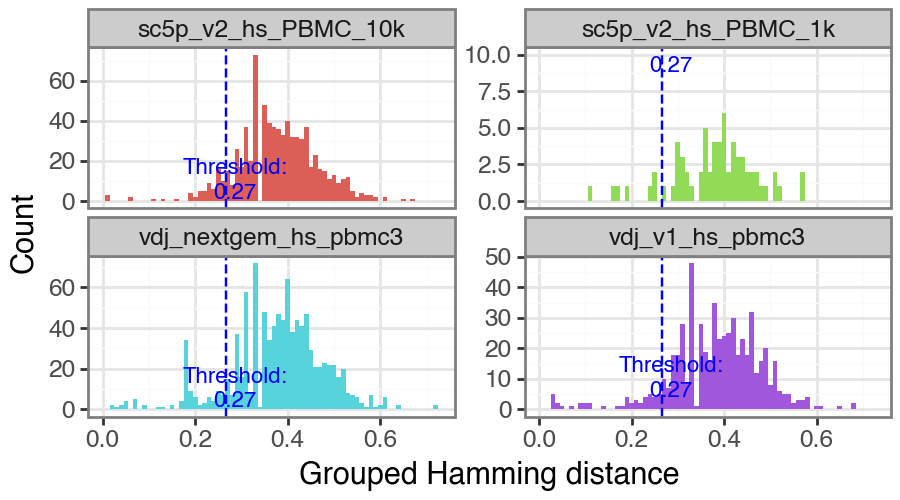
Alternatively, a wrapper to call changeo’s DefineClones.py [Gupta2015] is also included. To run it, you need to choose the distance threshold for clonal assignment. To facilitate this, the function pp.calculate_threshold will run shazam’s distToNearest function and return a plot showing the length normalized hamming distance distribution and
automated threshold value.
Again, pp.calculate_threshold will take a file path, pandas DataFrame or Dandelion object as input. If a Dandelion object is provided, the threshold value will be inserted into the .threshold slot. For more fine control, please use shazam’s distToNearest and changeo’s DefineClones.py functions directly.
[30]:
ddl.pp.calculate_threshold(vdj)
Calculating threshold
R[write to console]: Error in (function (db, sequenceColumn = "junction", vCallColumn = "v_call", :
361 cell(s) with multiple heavy chains found. One heavy chain per cell is expected.
Rerun this after filtering. For now, switching to heavy mode.
Threshold method 'density' did not return with any values. Switching to method = 'gmm'.
finished: Updated Dandelion object:
'threshold', threshold value for tuning clonal assignment
(0:01:24)
[31]:
# see the actual value in .threshold slot
vdj.threshold
[31]:
np.float64(0.26535373487471386)
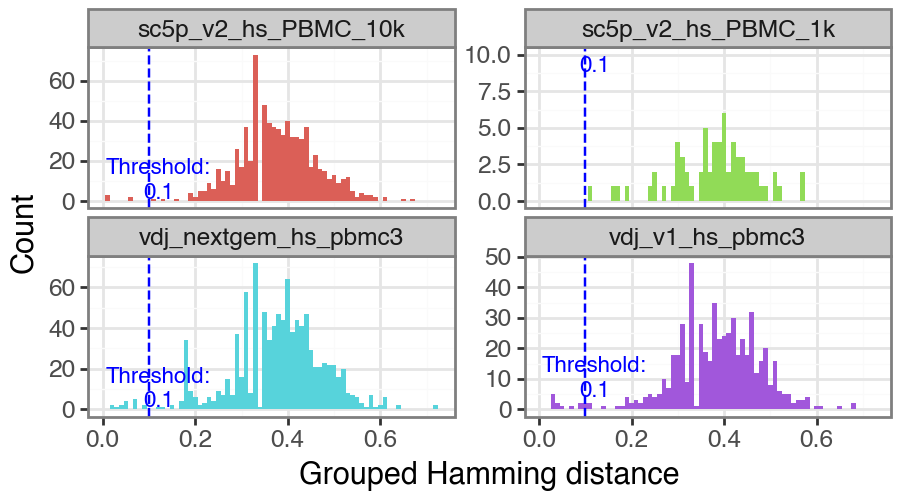
You can also manually select a value as the threshold if you wish. Note that rerunning this with manual_threshold is just for reproducing the plot but with the line at 0.1 in this tutorial. You can just edit vdj.threshold directly if you wish, i.e. vdj.threshold = 0.1.
[32]:
ddl.pp.calculate_threshold(vdj, manual_threshold=0.1)
Calculating threshold
R[write to console]: Error in (function (db, sequenceColumn = "junction", vCallColumn = "v_call", :
361 cell(s) with multiple heavy chains found. One heavy chain per cell is expected.
Rerun this after filtering. For now, switching to heavy mode.
finished: Updated Dandelion object:
'threshold', threshold value for tuning clonal assignment
(0:00:04)
[33]:
# see the updated .threshold slot
vdj.threshold
[33]:
0.1
We can run ddl.tl.define_clones to call changeo’s DefineClones.py; see here for more info. Note, if a pandas.DataFrame or file path is provided as the input, the value in dist option (corresponds to threshold value) needs to be manually supplied. If a Dandelion object is provided, it will automatically retrieve it from the threshold slot. Additional options for ddl.tl.define_clones to provide to
DefineClones.py can be supplied as a list to the additional_args option.
[34]:
ddl.tl.define_clones(vdj, key_added="changeo_clone_id")
vdj
Finding clones
Running command: DefineClones.py -d /var/folders/_r/j_8_fj3x28n2th3ch0ckn9c40000gt/T/tmpgr2ignig/tmp/dandelion_define_clones_heavy-clone.tsv -o /var/folders/_r/j_8_fj3x28n2th3ch0ckn9c40000gt/T/tmpgr2ignig/dandelion_define_clones_heavy-clone.tsv --act set --model ham --norm len --dist 0.1 --nproc 1 --vf v_call_genotyped
START> DefineClones
FILE> dandelion_define_clones_heavy-clone.tsv
SEQ_FIELD> junction
V_FIELD> v_call_genotyped
J_FIELD> j_call
MAX_MISSING> 0
GROUP_FIELDS> None
ACTION> set
MODE> gene
DISTANCE> 0.1
LINKAGE> single
MODEL> ham
NORM> len
SYM> avg
NPROC> 1
PROGRESS> [Grouping sequences] 16:11:14 (2178) 0.0 min
PROGRESS> [Assigning clones] 16:11:16 |####################| 100% (2,178) 0.0 min
OUTPUT> dandelion_define_clones_heavy-clone.tsv
CLONES> 2118
RECORDS> 2178
PASS> 2178
FAIL> 0
END> DefineClones
finished: Updated Dandelion object:
'data', contig-indexed AIRR table
'metadata', cell-indexed observations table
(0:00:07)
[34]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status', 'clone_id', 'changeo_clone_id'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'changeo_clone_id'
Note that I specified the option key_added and this adds the output from tl.define_clones into a separate column. If left as default (None), it will write into clone_id column. The same option can be specified in tl.find_clones earlier.
Other clustering methods
dandelion also now supports clustering methods from scoper. You can access the functions through:
dandelion.external.immcancation.scoper.identical_clones(vdj, ...)
dandelion.external.immcancation.scoper.hierarchical_clones(vdj, ...)
dandelion.external.immcancation.scoper.spectral_clones(vdj, ...)
see the dandelion documentation and original scoper documentation for more details.
As more methods arise, we will continue to evaluate which ones to include in dandelion.
Generation of V(D)J network
dandelion generates a network to facilitate visualisation of results, inspired from [Bashford-Rogers2013]. This uses the full V(D)J contig sequences instead of just the junctional sequences to chart a tree-like network for each clone. The actual visualization will be achieved through scanpy later.
ddl.tl.generate_network
First we need to generate the network. ddl.tl.generate_network will take a V(D)J table that has clones defined, specifically under the 'clone_id' column. The default mode is to use amino acid sequences for constructing Levenshtein distance matrices, but can be toggled using the key option.
If you have a pre-processed table parsed from immcantation’s method, or any other method as long as it’s in a AIRR format, the table can be used as well.
You can specify the clone_key option for generating the network for the clone id definition of choice as long as it exists as a column in the .data slot.
Before proceeding, let’s do a bit of subsetting. Here I want to remove the Orphan VJ cells (lacking BCR heavy chain i.e. VDJ information). Whether or not you want to do this is up to you. I’m doing this because I want to focus on the BCR heavy chain for now. You may elect to keep everything and that can be your starting point for further analysis.
[35]:
vdj = vdj[
vdj.metadata.chain_status.isin(
["Single pair", "Extra pair", "Extra pair-exception", "Orphan VDJ"]
)
].copy()
vdj
[35]:
Dandelion class object with n_obs = 2112 and n_contigs = 5052
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status', 'clone_id', 'changeo_clone_id'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'changeo_clone_id'
[36]:
ddl.tl.generate_network(vdj)
Generating network
Setting up data: 4424it [00:00, 7756.14it/s]
Calculating distances : 100%|██████████| 2313/2313 [00:00<00:00, 9523.15it/s]
Aggregating distances : 100%|██████████| 5/5 [00:00<00:00, 126.71it/s]
Sorting into clusters : 100%|██████████| 2313/2313 [00:00<00:00, 4563.84it/s]
Calculating minimum spanning tree : 100%|██████████| 35/35 [00:00<00:00, 1223.91it/s]
Generating edge list : 100%|██████████| 35/35 [00:00<00:00, 4550.12it/s]
Computing overlap : 100%|██████████| 2313/2313 [00:00<00:00, 2455.89it/s]
Adjust overlap : 100%|██████████| 143/143 [00:00<00:00, 4625.55it/s]
Linking edges : 100%|██████████| 2062/2062 [00:00<00:00, 52032.94it/s]
Computing network layout
Computing expanded network layout
finished: Updated Dandelion object:
'data', contig-indexed AIRR table
'metadata', cell-indexed observations table
'layout', graph layout
'graph', network constructed from distance matrices of VDJ- and VJ- chains (0:00:04)
In dandelion version >=0.2.2, the default layout_method is changed to sfdp, which is implemented through graph-tool package. This is significantly faster than the default modified Fruchterman-Reingold layout which while will work reasonably fast here, it will take quite a while when a lot of contigs are provided (>100k cells may take 1 hour). You can toggle this behaviour with:
ddl.tl.generate_network(vdj, layout_method = 'mod_fr') # for the original
ddl.tl.generate_network(vdj, layout_method = 'sfdp') # for sfdp
Generating graph without layout
If you don’t care for the layout and simply want access to the network/graph, you can do:
ddl.tl.generate_network(vdj, compute_layout = False)
and use the networkx graphs in the graph slot and compute your own layout as you wish.
In previous versions of dandelion, it used to be possible to generate the entire distance matrix for every pair of cell but this functionality was removed because it was too time consuming. If you are after this, please reach out to me and we can try and see if we can reimplement it!
down sampling data/graph
You can also downsample the number of cells. This will return a new object as a downsampled copy of the original with its own distance matrix. We will add use_existing_graph=False for this to work (otherwise it will just reuse the previous graph to recompute a layout; it will throw an error as it doesn’t know what to do with downsampling).
[37]:
vdj_downsample = ddl.tl.generate_network(
vdj, downsample=500, use_existing_graph=False
)
vdj_downsample
Generating network
Downsampling to 500 cells.
Setting up data: 1040it [00:00, 8537.89it/s]
Calculating distances : 100%|██████████| 549/549 [00:00<00:00, 15436.98it/s]
Aggregating distances : 100%|██████████| 4/4 [00:00<00:00, 1448.68it/s]
Sorting into clusters : 100%|██████████| 549/549 [00:00<00:00, 7272.88it/s]
Calculating minimum spanning tree : 100%|██████████| 3/3 [00:00<00:00, 987.51it/s]
Generating edge list : 100%|██████████| 3/3 [00:00<00:00, 3541.49it/s]
Computing overlap : 100%|██████████| 549/549 [00:00<00:00, 4610.85it/s]
Adjust overlap : 100%|██████████| 33/33 [00:00<00:00, 4357.65it/s]
Linking edges : 100%|██████████| 494/494 [00:00<00:00, 126117.61it/s]
Computing network layout
Computing expanded network layout
finished: Updated Dandelion object:
'data', contig-indexed AIRR table
'metadata', cell-indexed observations table
'layout', graph layout
'graph', network constructed from distance matrices of VDJ- and VJ- chains (0:00:01)
[37]:
Dandelion class object with n_obs = 500 and n_contigs = 1040
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status', 'clone_id', 'changeo_clone_id'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'changeo_clone_id'
layout: layout for 500 vertices, layout for 9 vertices
graph: networkx graph of 500 vertices, networkx graph of 9 vertices
check the newly re-initialized Dandelion object
[38]:
vdj
[38]:
Dandelion class object with n_obs = 2112 and n_contigs = 5052
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status', 'clone_id', 'changeo_clone_id'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'changeo_clone_id'
layout: layout for 2112 vertices, layout for 71 vertices
graph: networkx graph of 2112 vertices, networkx graph of 71 vertices
The graph/networks can be accessed through the .graph slot as an networkx graph object if you want to extract the data for network statistics or make any changes to the network.
At this point, we can save the dandelion object.
[39]:
vdj.write_h5ddl("dandelion_results_simplified.h5ddl", compression="gzip")
[40]:
# let's also transfer the new slots to the original files
vdj_original = ddl.read_h5ddl("dandelion_results.h5ddl")
vdj_original
[40]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status'
metadata: 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
[41]:
vdj_original.metadata = vdj.metadata.copy()
vdj_original.layout = vdj.layout
vdj_original.graph = vdj.graph
vdj_original
[41]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status'
metadata: 'clone_id', 'clone_id_by_size', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ', 'changeo_clone_id'
layout: layout for 2112 vertices, layout for 71 vertices
graph: networkx graph of 2112 vertices, networkx graph of 71 vertices
[42]:
# save
vdj_original.write_h5ddl("dandelion_results.h5ddl", compression="gzip")
[ ]: