Filtering
We now move on to filtering out BCR contigs (and corresponding cells if necessary) from the BCR data and transcriptome object loaded in scanpy.
Import dandelion module
[1]:
import os
import dandelion as ddl
# change directory to somewhere more workable
os.chdir(os.path.expanduser("~/Downloads/dandelion_tutorial/"))
ddl.logging.print_header()
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/anndata/utils.py:429: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
dandelion==0.5.5.dev16 pandas==2.2.3 numpy==2.1.3 matplotlib==3.10.1 networkx==3.4.2 scipy==1.15.2
/opt/homebrew/Caskroom/miniforge/base/envs/dandelion/lib/python3.11/site-packages/nxviz/__init__.py:33: UserWarning:
nxviz has a new API! Version 0.7.4 onwards, the old class-based API is being
deprecated in favour of a new API focused on advancing a grammar of network
graphics. If your plotting code depends on the old API, please consider
pinning nxviz at version 0.7.4, as the new API will break your old code.
To check out the new API, please head over to the docs at
https://ericmjl.github.io/nxviz/ to learn more. We hope you enjoy using it!
(This deprecation message will go away in version 1.0.)
Import modules for use with scanpy
[2]:
import pandas as pd
import scanpy as sc
import warnings
warnings.filterwarnings("ignore")
sc.logging.print_header()
scanpy==1.10.3 anndata==0.11.3 umap==0.5.7 numpy==2.1.3 scipy==1.15.2 pandas==2.2.3 scikit-learn==1.6.1 statsmodels==0.14.4 igraph==0.11.8 pynndescent==0.5.13
Import the transcriptome data
[3]:
samples = [
"sc5p_v2_hs_PBMC_1k",
"sc5p_v2_hs_PBMC_10k",
"vdj_v1_hs_pbmc3",
"vdj_nextgem_hs_pbmc3",
]
adata_list = []
for sample in samples:
adata = sc.read_10x_h5(
sample + "/filtered_feature_bc_matrix.h5", gex_only=True
)
adata.obs["sampleid"] = sample
# rename cells to sample id + barcode
adata.obs_names = [str(sample) + "_" + str(j) for j in adata.obs_names]
adata.var_names_make_unique()
adata_list.append(adata)
adata = adata_list[0].concatenate(adata_list[1:])
# rename the obs_names again, this time cleaving the trailing -#
adata.obs_names = [str(j).split("-")[0] for j in adata.obs_names]
adata
[3]:
AnnData object with n_obs × n_vars = 30471 × 31915
obs: 'sampleid', 'batch'
var: 'feature_types', 'genome', 'pattern', 'read', 'sequence', 'gene_ids-0', 'gene_ids-1', 'gene_ids-2', 'gene_ids-3'
I’m using a wrapper called pp.recipe_scanpy_qc to run through a generic scanpy workflow. You can skip this if you already have a pre-processed AnnData object for the subsequent steps.
[4]:
ddl.pp.recipe_scanpy_qc(adata, mito_cutoff=None) # use a gmm model to decide
# we can continue with those that survive qc
adata = adata[adata.obs["filter_rna"] == "False"].copy()
adata
OMP: Info #276: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
[4]:
AnnData object with n_obs × n_vars = 23715 × 31915
obs: 'sampleid', 'batch', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'gmm_pct_count_clusters_keep', 'scrublet_score', 'is_doublet', 'filter_rna'
var: 'feature_types', 'genome', 'pattern', 'read', 'sequence', 'gene_ids-0', 'gene_ids-1', 'gene_ids-2', 'gene_ids-3'
Filter cells that are potental doublets and poor quality in both the V(D)J data and transcriptome data
ddl.pp.check_contigs
Deprecation warning
Pre v0.5.0, there are two separate functions to perform contig QC with either ddl.pp.filter_contigs or ddl.pp.check_contigs to deal with poor quality contigs, either explicitly removing them or just flagging them. From v0.5.0 onwards however, ddl.pp.filter_contigs is deprecated and will be removed in v0.6.0, and ddl.pp.check_contigs will be the only QC option going forward. ddl.pp.check_contigs is easier to maintain and simply marks the problematic contigs as ambiguous
and withhold them from downstream analysis. The new version of ddl.pp.check_contigs will also have the filter_extra and filter_ambiguous options to remove/keep the extra (marked due to passing the internal QC filters but not explicitly ambiguous) and ambiguous contigs, fulfilling the same utility as ddl.pp.filter_contigs.
We use the function pp.check_contigs to mark and filter out cells and contigs from both the V(D)J data and transcriptome data in AnnData. The operation will remove bad quality cells based on transcriptome information as well as remove V(D)J doublets (multiplet heavy/long chains, and/or light/short chains) from the V(D)J data. In some situations, a single cell can have multiple heavy/long and light/short chain contigs although they have an identical V(D)J+C alignment; in situations like
this, the contigs with lesser UMIs will be dropped and the UMIs transferred to umi_count column. The same procedure is applied to both chains before further checks of the annotation quality, UMI and consensus count distributions.
Cells in the gene expression object without V(D)J information will not be affected which means that the AnnData object can hold non-B/T cells.
[5]:
# first we read in the 4 bcr files
bcr_files = []
for sample in samples:
file_location = sample + "/dandelion/filtered_contig_dandelion.tsv"
bcr_files.append(pd.read_csv(file_location, sep="\t"))
bcr = pd.concat(bcr_files, ignore_index=True)
bcr.reset_index(inplace=True, drop=True)
bcr
[5]:
| sequence_id | sequence | rev_comp | productive | v_call | d_call | j_call | sequence_alignment | germline_alignment | junction | ... | v_sequence_alignment_aa | d_sequence_alignment_aa | j_sequence_alignment_aa | complete_vdj | j_call_multimappers | j_call_multiplicity | j_call_sequence_start_multimappers | j_call_sequence_end_multimappers | j_call_support_multimappers | mu_count | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | sc5p_v2_hs_PBMC_1k_AACTCCCAGGCTAGGT_contig_1 | ACTGCGGGGGTAAGAGGTTGTGTCCACCATGGCCTGGACTCCTCTC... | F | T | IGLV5-45*03 | NaN | IGLJ3*02 | CAGGCTGTGCTGACTCAGCCGTCTTCC...CTCTCTGCATCTCCTG... | CAGGCTGTGCTGACTCAGCCGTCTTCC...CTCTCTGCATCTCCTG... | TGTATGATTTGGCACAGCAGCGCTTGGGTGTTC | ... | QAVLTQPSSLSASPGASASLTCTLRSGINVGTYRIYWYQQKPGSPP... | NaN | VFGGGTKLTVL | NaN | IGLJ3*01 | 1.0 | 397 | 431 | 0.0 | 0 |
| 1 | sc5p_v2_hs_PBMC_1k_AACTCCCAGGCTAGGT_contig_2 | ATACTTTCTGAGAGTCCTGGACCTCCTGTGCAAGAACATGAAACAT... | F | T | IGHV4-61*02 | IGHD3-3*01 | IGHJ6*02 | CAGGTGCAGCTGCAGGAGTCGGGCCCA...GGACTGGTGAAGCCTT... | CAGGTGCAGCTGCAGGAGTCGGGCCCA...GGACTGGTGAAGCCTT... | TGTGCGAGAGAAAATTACGATTTTTGGAGTGGTTATTACCACGGTG... | ... | QVQLQESGPGLVKPSQTLSLTCTVSGGSISSGSYYWSWIRQPAGKG... | YDFWSGY | YHGADVWGQGTTVTVSS | NaN | IGHJ6*02 | 1.0 | 416 | 470 | 0.0 | 3 |
| 2 | sc5p_v2_hs_PBMC_1k_AACTCCCAGGCTAGGT_contig_3 | GGCTGGGGTCTCAGGAGGCAGCGCTCTGGGGACGTCTCCACCATGG... | F | F | IGLV2-5*01 | NaN | IGLJ3*02 | CAGTCTGCCCTGATTCAGCCTCCCTCC...GTGTCCGGGTCTCCTG... | CAGTCTGCCCTGATTCAGCCTCCCTCC...GTGTCCGGGTCTCCTG... | TGCTGCTCATATACAAGCAGTGCCACTTTCTTGGGTGTTC | ... | QSALIQPPSVSGSPGQSVTISCTGTSSDVGSYDYVSWYQQHPGTVP... | NaN | LGVRRRDQADRP | NaN | IGLJ3*02 | 1.0 | 396 | 433 | 0.0 | 0 |
| 3 | sc5p_v2_hs_PBMC_1k_AACTCTTGTCATCGGC_contig_2 | AGCTCTGAGAGAGGAGCCTTAGCCCTGGATTCCAAGGCCTATCCAC... | F | T | IGHV3-21*01 | IGHD3-22*01 | IGHJ4*02 | GAGGTGCAGCTGGTGGAGTCTGGGGGA...GGCCTGGTCAAGCCTG... | GAGGTGCAGCTGGTGGAGTCTGGGGGA...GGCCTGGTCAAGCCTG... | TGTGCGAGACGTTACTATGATAGTAGTGGTTATTCCGCAAACTTTG... | ... | EVQLVESGGGLVKPGGSLRLSCAASGFTFSSYSMNWVRQAPGKGLE... | YYDSSGY | FDYWGQGTLVTVSS | NaN | IGHJ4*02 | 1.0 | 462 | 506 | 0.0 | 0 |
| 4 | sc5p_v2_hs_PBMC_1k_AACTCTTGTCATCGGC_contig_1 | AGAGCTCTGGGGAGTCTGCACCATGGCTTGGACCCCACTCCTCTTC... | F | T | IGLV4-69*01 | NaN | IGLJ1*01 | CAGCTTGTGCTGACTCAATCGCCCTCT...GCCTCTGCCTCCCTGG... | CAGCTTGTGCTGACTCAATCGCCCTCT...GCCTCTGCCTCCCTGG... | TGTCAGACCTGGGGCACTGGCATTTATGTCTTC | ... | QLVLTQSPSASASLGASVKLTCTLSSGHSSYAIAWHQQQPEKGPRY... | NaN | YVFGTGTKVTVL | NaN | IGLJ1*01 | 1.0 | 379 | 416 | 0.0 | 0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 7350 | vdj_nextgem_hs_pbmc3_TTTGCGCTCTGTCAAG_contig_2 | ATCACATAACAACCACATTCCTCCTCTAAAGAAGCCCCCGGGAGCC... | F | T | IGHV1-69*01,IGHV1-69D*01 | IGHD3-22*01 | IGHJ4*02 | CAGGTGCAGCTGGTGCAGTCTGGGGCT...GAAGTGAAGAAGCCTG... | CAGGTGCAGCTGGTGCAGTCTGGGGCT...GAGGTGAAGAAGCCTG... | TGTGCGAGGGGGAAGTATTACTATGATAAAAGTGGGTCTCCACCTC... | ... | QVQLVQSGAEVKKPGSSVKVSCKVSGGIFSSYAISWVRQAPGQGLE... | YYYDKSG | FDYWGQGTLVTVSS | NaN | IGHJ4*02 | 1.0 | 457 | 500 | 1.18e-19 | 16 |
| 7351 | vdj_nextgem_hs_pbmc3_TTTGGTTGTAAGGATT_contig_1 | AGAGCTCTGGAGAAGAGCTGCTCAGTTAGGACCCAGAGGGAACCAT... | F | T | IGKV3-20*01 | NaN | IGKJ2*01,IGKJ2*02 | GAAATTGTGTTGACGCAGTCTCCAGGCACCCTGTCTTTGTCTCCAG... | GAAATTGTGTTGACGCAGTCTCCAGGCACCCTGTCTTTGTCTCCAG... | TGTCAGCAGTATGATGAGTCACCTCTGACTTTT | ... | EIVLTQSPGTLSLSPGERATLSCRASQSLTNSQLAWYQQKPGQAPR... | NaN | TFGQGTKLEIK | NaN | IGKJ2*02 | 1.0 | 396 | 429 | 3.59e-14 | 11 |
| 7352 | vdj_nextgem_hs_pbmc3_TTTGGTTGTAAGGATT_contig_2 | AGCTCTGGGAGAGGAGCCCCAGCCCTGAGATTCCCAGGTGTTTCCA... | F | T | IGHV3-9*01 | IGHD5-18*01,IGHD5-5*01 | IGHJ6*03 | GAAGTGCAGCTGGTGGAGTCTGGGGGA...GGCTTGGTACAGCCTG... | GAAGTGCAGCTGGTGGAGTCTGGGGGA...GGCTTGGTACAGCCTG... | TGTGCAAAAGACGGATACAGCTATCGTTCGTCATACTACTTTTACA... | ... | EVQLVESGGGLVQPGRSLRLSCAASGFSFDDYVMHWVRQAPGKGLE... | GYSYR | YYFYMDVWGKGTTVTVSS | NaN | IGHJ6*03 | 1.0 | 456 | 510 | 1.96e-22 | 10 |
| 7353 | vdj_nextgem_hs_pbmc3_TTTGTCACAGTAGAGC_contig_1 | AGCTCTGAGAGAGGAGCCCAGCCCTGGGATTTTCAGGTGTTTTCAT... | F | T | IGHV3-23*01,IGHV3-23D*01 | IGHD4-17*01 | IGHJ4*02 | GAGGTGCAGCTGTTGGAGTCTGGGGGA...GGCTTGGTACAGCCTG... | GAGGTGCAGCTGTTGGAGTCTGGGGGA...GGCTTGGTACAGCCTG... | TGTGCGAAAGATTTTAGGTCGCCATACGGTGACTACTACTTTGACT... | ... | EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYAMSWVRQAPGKGLE... | YGD | YFDYWGQGTLVTVSS | NaN | IGHJ4*02 | 1.0 | 456 | 503 | 6.02e-22 | 0 |
| 7354 | vdj_nextgem_hs_pbmc3_TTTGTCACAGTAGAGC_contig_2 | GTGGGTCCAGGAGGCAGAACTCTGGGTGTCTCACCATGGCCTGGAT... | F | T | IGLV3-25*03 | NaN | IGLJ1*01 | TCCTATGAGCTGACACAGCCACCCTCG...GTGTCAGTGTCCCCAG... | TCCTATGAGCTGACACAGCCACCCTCG...GTGTCAGTGTCCCCAG... | TGTCAATCAGCAGACAGCAGTGGTACTTATCTTTATGTCTTC | ... | SYELTQPPSVSVSPGQTARITCSGDALPKQYAYWYQQKPGQAPVLV... | NaN | YVFGTGTKVTVL | NaN | IGLJ1*01 | 1.0 | 383 | 420 | 1.64e-16 | 0 |
7355 rows × 120 columns
Library type
It is recommended to specify the library_type argument as it will remove all contigs that do not belong to the related loci. The rationale is that the choice of the library type should mean that the primers used would most likely amplify those related sequences and if there’s any unexpected loci, they likely represent artifacts and shouldn’t be analysed. The optional argument accepts: ig, tr-ab, tr-gd or None where None means all contigs will be kept.
The main output of this function are two an additional columns in vdj.data, extra and ambiguous, which flags T or F for contigs that were marked accordingly. The rules for marking contigs are as follows:
extra is marked as T if the contig passes the internal QC filters based on umi_count (or consensus_count if there are ties in the umi_count) in a cell. If you are only interested in just the top contig pair, you can set filter_extra=True to remove the extra contigs.
For VDJ chains, the current rule set is to keep the top 1 productive contig with the highest counts and mark the rest as extra (or ambiguous if appropriate). Toggle ntop_vdj to keep the top n (default 1) contigs.
For VJ chains, the current rule set is to keep the top 2 productive contigs with the highest counts and mark the rest as extra (or ambiguous if appropriate). Toggle ntop_vj to keep the top n (default 2) contigs.
ambiguous is marked as T if the contig is of poor quality annotation and would be removed from downstream analysis. Cells with multiple contigs with very low umi_counts and/or consensus_counts are also marked as ambiguous as it is not possible to distinguish which is the most representative contig.
Please note that the default for filter_extra is True. If you want to keep the extra contigs for whatever reasons e.g. interested in T/B-cell development datasets, you need to set filter_extra=False. We are setting this as False in this example because later on we want to visualise these extra contigs.
```python
[6]:
vdj, adata = ddl.pp.check_contigs(
bcr, adata, library_type="ig", filter_extra=False
)
Preparing data: 6503it [00:00, 7804.71it/s]
Scanning for poor quality/ambiguous contigs: 100%|██████████| 3158/3158 [00:05<00:00, 559.36it/s]
Check the Dandelion object
[7]:
vdj
[7]:
Dandelion class object with n_obs = 2238 and n_contigs = 7355
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status'
metadata: 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
Check the AnnData object as well
[8]:
adata
[8]:
AnnData object with n_obs × n_vars = 23715 × 31915
obs: 'sampleid', 'batch', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'gmm_pct_count_clusters_keep', 'scrublet_score', 'is_doublet', 'filter_rna', 'has_contig', 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
var: 'feature_types', 'genome', 'pattern', 'read', 'sequence', 'gene_ids-0', 'gene_ids-1', 'gene_ids-2', 'gene_ids-3'
These are the relevant columns for looking at the QC status of the cells and contigs in the .obs slot in the AnnData object (and also .metadata slot in the Dandelion object):
Relevant columns in obs
has_contigwhether cells have V(D)J chains.
locus_statusdetailed information on chain status pairings (below).
chain_statussummarised information of the chain locus status pairings (similar to
chain_pairinginscirpy).rearrangement_status_VDJandrearrangement_status_VJwhether or not V(D)J gene usage are standard (i.e. all from the same locus).
So in a standard situation, I would remove cells flagged with Orphan VJ, Orphan VJ-exception, ambiguous in .metadata.chain_status, and also any cell marked as chimeric in the .metadata.rearrangement_status_VDJ and .metadata.rearrangement_status_VJ from downstream cell-level calculations/analysis.
Having said that, you will find that most of Dandelion’s functions will work without the need to requirement to perform additional filtering and filtering can be performed on the final AnnData object (described in the visualisation section).
Let’s take a look at these new columns
[9]:
pd.crosstab(adata.obs["chain_status"], adata.obs["locus_status"])
[9]:
| locus_status | Extra VDJ + Extra VJ | Extra VDJ + IGK | Extra VDJ + IGL | IGH + Extra VJ | IGH + IGK | IGH + IGL | No_contig | Orphan Extra VJ | Orphan IGH | Orphan IGK | Orphan IGL |
|---|---|---|---|---|---|---|---|---|---|---|---|
| chain_status | |||||||||||
| Extra pair | 58 | 4 | 4 | 84 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| No_contig | 0 | 0 | 0 | 0 | 0 | 0 | 21477 | 0 | 0 | 0 | 0 |
| Orphan Extra VJ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| Orphan VDJ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 |
| Orphan VJ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 84 | 39 |
| Single pair | 0 | 0 | 0 | 0 | 1129 | 824 | 0 | 0 | 0 | 0 | 0 |
if there are multiple library types, i.e. ddl.pp.filter_contigs or ddl.pp.check_contigs was run with library_type = None, or if several tcr/bcr Dandelion objects are concatenated, there will be additional columns where the v/d/j/c calls and productive will be split into additional columns to reflect those that belong to a B cell, alpha-beta T cell, or gamma-delta T cell.
We will use this contig_checked object going forward.
Now actually filter the AnnData object and run through a standard workflow starting by filtering genes and normalizing the data
Because the ‘filtered’ AnnData object was returned as a filtered but otherwise unprocessed object, we still need to normalize and run through the usual process here. The following is just a standard scanpy workflow.
[10]:
# filter genes
sc.pp.filter_genes(adata, min_cells=3)
# Normalize the counts
sc.pp.normalize_total(adata, target_sum=1e4)
# Logarithmize the data
sc.pp.log1p(adata)
# Stash the normalised counts
adata.raw = adata
Identify highly-variable genes
[11]:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)
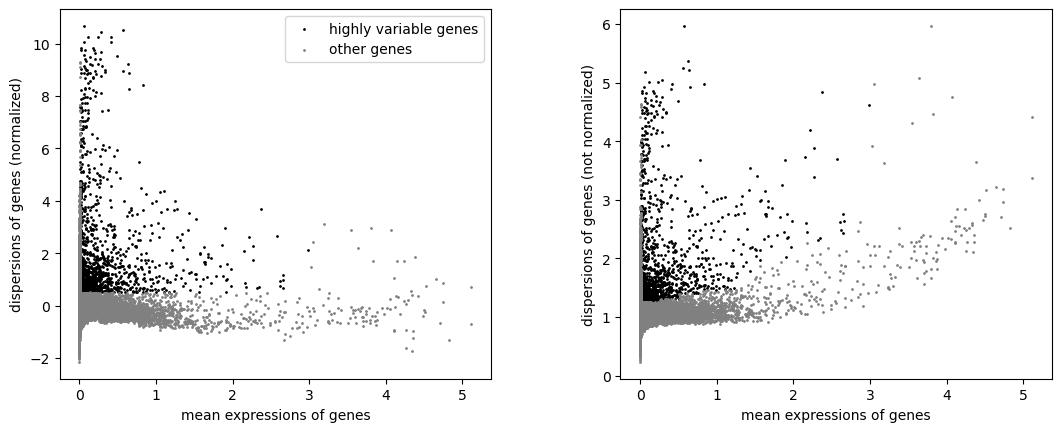
Filter the genes to only those marked as highly-variable
[12]:
adata = adata[:, adata.var.highly_variable]
Regress out effects of total counts per cell and the percentage of mitochondrial genes expressed. Scale the data to unit variance.
[13]:
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"])
sc.pp.scale(adata, max_value=10)
Run PCA
[14]:
sc.tl.pca(adata, svd_solver="arpack")
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=50)
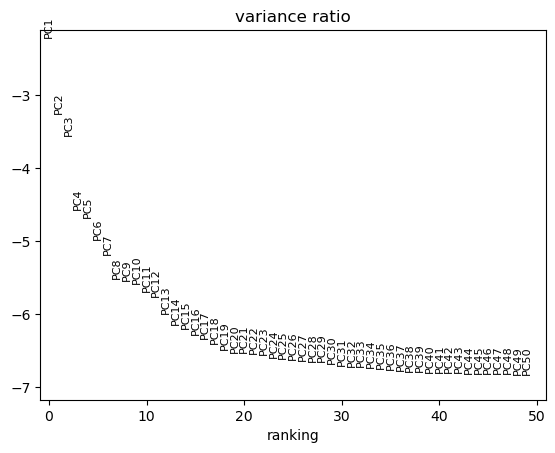
Computing the neighborhood graph, umap and clusters
[15]:
# Computing the neighborhood graph
sc.pp.neighbors(adata)
# Embedding the neighborhood graph
sc.tl.umap(adata)
# Clustering the neighborhood graph
sc.tl.leiden(adata)
Visualizing the clusters and whether or not there’s a corresponding V(D)J receptor
[16]:
sc.pl.umap(adata, color=["leiden", "chain_status"])
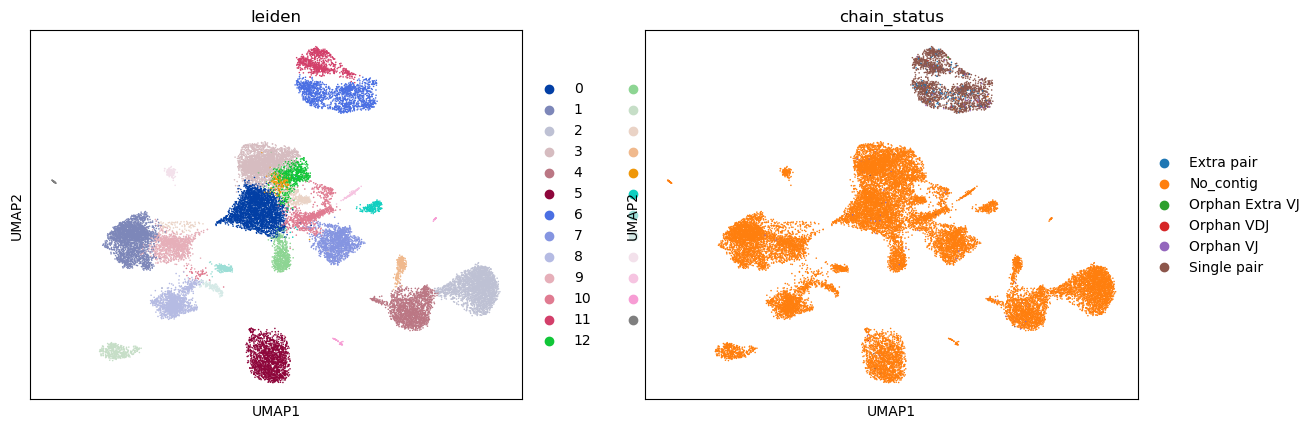
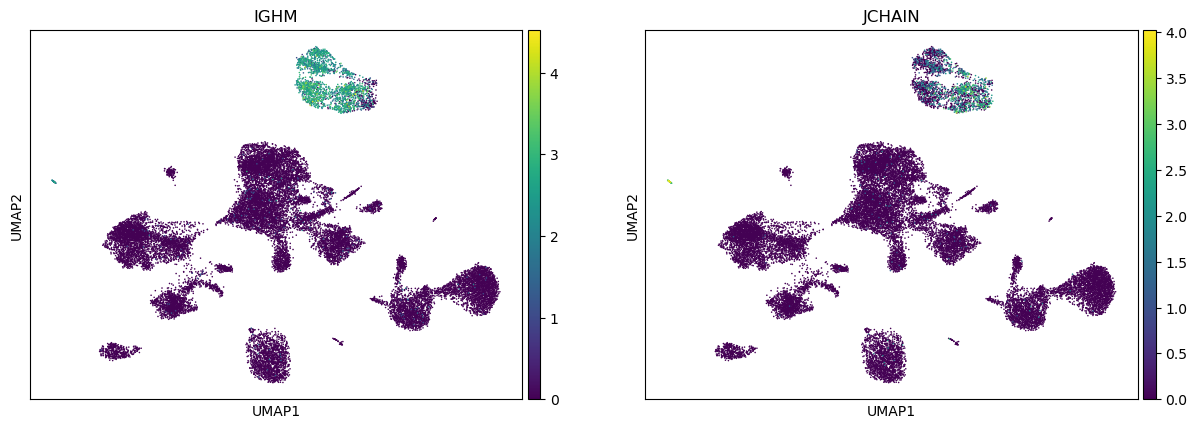
Visualizing some B cell genes
[17]:
sc.pl.umap(adata, color=["IGHM", "JCHAIN"])
Save AnnData
We can save this AnnData object for now.
[18]:
adata.write("adata.h5ad", compression="gzip")
Save dandelion
To save the vdj object, we have two options - either save the .data and .metadata slots with pandas’ functions:
[19]:
vdj.data.to_csv("filtered_vdj_table.tsv", sep="\t")
Or save the whole Dandelion class object with either .write_h5ddl/.write, which saves the class to a HDF5 format, or using a pickle-based .write_pkl function.
From v0.4.0, the .write_h5ddl/.write function has been refactored to use h5py. Support for files saved prior to v0.4.0 (which used pandas to save in HDF5 format) will be maintained at least until the next major version and ddl.read_h5ddl will be able to read both old and new versions. The old version can be saved with .write_h5ddl(..., version=3) but this is not covered by tests because of issues with installing the dependencies.
[20]:
vdj.write_h5ddl("dandelion_results.h5ddl") # can add compression="gzip"
[21]:
vdj.write_pkl(
"dandelion_results.pkl.pbz2"
) # this will automatically use bzip2 for compression, switch the extension to .gz for gzip
Running ddl.pp.check_contigs without AnnData
Finally, ddl.pp.check_contigs can also be run without an AnnData object:
[22]:
vdj3 = ddl.pp.check_contigs(bcr)
vdj3
Preparing data: 6503it [00:00, 7410.51it/s]
Scanning for poor quality/ambiguous contigs: 100%|██████████| 3158/3158 [00:06<00:00, 524.58it/s]
[22]:
Dandelion class object with n_obs = 3158 and n_contigs = 6374
data: 'sequence_id', 'sequence', 'rev_comp', 'productive', 'v_call', 'd_call', 'j_call', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'v_cigar', 'd_cigar', 'j_cigar', 'stop_codon', 'vj_in_frame', 'locus', 'c_call', 'junction_length', 'np1_length', 'np2_length', 'v_sequence_start', 'v_sequence_end', 'v_germline_start', 'v_germline_end', 'd_sequence_start', 'd_sequence_end', 'd_germline_start', 'd_germline_end', 'j_sequence_start', 'j_sequence_end', 'j_germline_start', 'j_germline_end', 'v_score', 'v_identity', 'v_support', 'd_score', 'd_identity', 'd_support', 'j_score', 'j_identity', 'j_support', 'fwr1', 'fwr2', 'fwr3', 'fwr4', 'cdr1', 'cdr2', 'cdr3', 'cell_id', 'consensus_count', 'umi_count', 'v_call_10x', 'd_call_10x', 'j_call_10x', 'junction_10x', 'junction_10x_aa', 'j_support_igblastn', 'j_score_igblastn', 'j_call_igblastn', 'j_call_blastn', 'j_identity_blastn', 'j_alignment_length_blastn', 'j_number_of_mismatches_blastn', 'j_number_of_gap_openings_blastn', 'j_sequence_start_blastn', 'j_sequence_end_blastn', 'j_germline_start_blastn', 'j_germline_end_blastn', 'j_support_blastn', 'j_score_blastn', 'j_sequence_alignment_blastn', 'j_germline_alignment_blastn', 'j_source', 'd_support_igblastn', 'd_score_igblastn', 'd_call_igblastn', 'd_call_blastn', 'd_identity_blastn', 'd_alignment_length_blastn', 'd_number_of_mismatches_blastn', 'd_number_of_gap_openings_blastn', 'd_sequence_start_blastn', 'd_sequence_end_blastn', 'd_germline_start_blastn', 'd_germline_end_blastn', 'd_support_blastn', 'd_score_blastn', 'd_sequence_alignment_blastn', 'd_germline_alignment_blastn', 'd_source', 'v_call_genotyped', 'germline_alignment_d_mask', 'sample_id', 'c_sequence_alignment', 'c_germline_alignment', 'c_sequence_start', 'c_sequence_end', 'c_score', 'c_identity', 'c_call_10x', 'junction_aa_length', 'fwr1_aa', 'fwr2_aa', 'fwr3_aa', 'fwr4_aa', 'cdr1_aa', 'cdr2_aa', 'cdr3_aa', 'sequence_alignment_aa', 'v_sequence_alignment_aa', 'd_sequence_alignment_aa', 'j_sequence_alignment_aa', 'complete_vdj', 'j_call_multimappers', 'j_call_multiplicity', 'j_call_sequence_start_multimappers', 'j_call_sequence_end_multimappers', 'j_call_support_multimappers', 'mu_count', 'ambiguous', 'extra', 'rearrangement_status'
metadata: 'sample_id', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_genotyped_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_genotyped_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_genotyped_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_genotyped_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'umi_count_B_VDJ', 'umi_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
[ ]: