Reading 10X Cell Ranger output directly
If for whatever reason you’ve decided to skip the reannotation/preprocessing, you can read the files directly from the Cell Ranger output folder with Dandelion’s ddl.read_10x_vdj, which accepts the *_contig_annotations.csv or all_contig_annotations.json file(s) as input. If reading with the .csv file, and the .fasta file and/or .json file(s) are in the same folder, ddl.read_10x_vdj will try to extract additional information not found in the .csv file e.g.
contig sequences.
From Cell Ranger V4 onwards, there is also an airr_rearrangement.tsv file that can be used directly with Dandelion. However, doing so will miss out on the reannotation steps but that is entirely up to you.
We will download the airr_rearrangement.tsv file from here:
# bash
wget https://cf.10xgenomics.com/samples/cell-vdj/4.0.0/sc5p_v2_hs_PBMC_10k/sc5p_v2_hs_PBMC_10k_b_filtered_contig_annotations.csv
wget https://cf.10xgenomics.com/samples/cell-vdj/4.0.0/sc5p_v2_hs_PBMC_10k/sc5p_v2_hs_PBMC_10k_b_filtered_contig.fasta
# wget https://cf.10xgenomics.com/samples/cell-vdj/4.0.0/sc5p_v2_hs_PBMC_10k/sc5p_v2_hs_PBMC_10k_b_all_contig_annotations.json
wget https://cf.10xgenomics.com/samples/cell-vdj/4.0.0/sc5p_v2_hs_PBMC_10k/sc5p_v2_hs_PBMC_10k_b_airr_rearrangement.tsv
Import dandelion module
[1]:
import os
import dandelion as ddl
# change directory to somewhere more workable
os.chdir(os.path.expanduser("~/Downloads/dandelion_tutorial/"))
ddl.logging.print_versions()
dandelion==0.3.4.dev30 pandas==2.0.1 numpy==1.24.3 matplotlib==3.7.1 networkx==3.1 scipy==1.11.2
With ddl.read_10x_vdj:
[2]:
folder_location = "sc5p_v2_hs_PBMC_10k"
# or file_location = 'sc5p_v2_hs_PBMC_10k/'
vdj = ddl.read_10x_vdj(
folder_location, filename_prefix="sc5p_v2_hs_PBMC_10k_b_filtered"
)
vdj
[2]:
Dandelion class object with n_obs = 994 and n_contigs = 2601
data: 'cell_id', 'is_cell_10x', 'sequence_id', 'high_confidence_10x', 'sequence_length_10x', 'locus', 'v_call', 'd_call', 'j_call', 'c_call', 'complete_vdj', 'productive', 'junction_aa', 'junction', 'consensus_count', 'duplicate_count', 'clone_id', 'raw_consensus_id_10x', 'sequence', 'rearrangement_status'
metadata: 'clone_id', 'clone_id_by_size', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'duplicate_count_B_VDJ', 'duplicate_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
With ddl.read_10x_airr:
[3]:
# read in the airr_rearrangement.tsv file
file_location = (
"sc5p_v2_hs_PBMC_10k/sc5p_v2_hs_PBMC_10k_b_airr_rearrangement.tsv"
)
vdj = ddl.read_10x_airr(file_location)
vdj
[3]:
Dandelion class object with n_obs = 994 and n_contigs = 2093
data: 'cell_id', 'sequence_id', 'sequence', 'sequence_aa', 'productive', 'rev_comp', 'v_call', 'v_cigar', 'd_call', 'd_cigar', 'j_call', 'j_cigar', 'c_call', 'c_cigar', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'junction_length', 'junction_aa_length', 'v_sequence_start', 'v_sequence_end', 'd_sequence_start', 'd_sequence_end', 'j_sequence_start', 'j_sequence_end', 'c_sequence_start', 'c_sequence_end', 'consensus_count', 'duplicate_count', 'is_cell', 'locus', 'rearrangement_status'
metadata: 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'duplicate_count_B_VDJ', 'duplicate_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
We will continue with the rest of the filtering part of the analysis to show how it slots smoothly with the rest of the workflow.
Import modules for use with scanpy
[4]:
import pandas as pd
import numpy as np
import scanpy as sc
import warnings
import functools
import seaborn as sns
import scipy.stats
import anndata
warnings.filterwarnings("ignore")
sc.logging.print_header()
scanpy==1.9.3 anndata==0.9.1 umap==0.5.3 numpy==1.24.3 scipy==1.11.2 pandas==2.0.1 scikit-learn==1.3.0 statsmodels==0.14.0 python-igraph==0.10.6 pynndescent==0.5.10
Import the transcriptome data
[5]:
adata = sc.read_10x_h5(
"sc5p_v2_hs_PBMC_10k/filtered_feature_bc_matrix.h5", gex_only=True
)
adata.obs["sample_id"] = "sc5p_v2_hs_PBMC_10k"
adata.var_names_make_unique()
adata
[5]:
AnnData object with n_obs × n_vars = 10553 × 36601
obs: 'sample_id'
var: 'gene_ids', 'feature_types', 'genome'
Run QC on the transcriptome data.
[6]:
ddl.pp.recipe_scanpy_qc(adata)
adata
[6]:
AnnData object with n_obs × n_vars = 10553 × 36601
obs: 'sample_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'scrublet_score', 'is_doublet', 'filter_rna'
var: 'gene_ids', 'feature_types', 'genome'
Run the filtering of bcr data. Note that I’m using the Dandelion object as input rather than the pandas dataframe (yes both types of input will works. In fact, a file path to the .tsv will work too).
[7]:
# The function will return both objects.
vdj, adata = ddl.pp.check_contigs(vdj, adata)
Preparing data: 2093it [00:00, 16146.33it/s]
Scanning for poor quality/ambiguous contigs: 100%|██████████| 994/994 [00:01<00:00, 977.40it/s]
Check the output V(D)J table
The vdj table is returned as a Dandelion class object in the .data slot; if a file was provided for filter_bcr above, a new file will be created in the same folder with the filtered prefix. Note that this V(D)J table is indexed based on contigs (sequence_id).
[8]:
vdj
[8]:
Dandelion class object with n_obs = 984 and n_contigs = 2093
data: 'cell_id', 'sequence_id', 'sequence', 'sequence_aa', 'productive', 'rev_comp', 'v_call', 'v_cigar', 'd_call', 'd_cigar', 'j_call', 'j_cigar', 'c_call', 'c_cigar', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'junction_length', 'junction_aa_length', 'v_sequence_start', 'v_sequence_end', 'd_sequence_start', 'd_sequence_end', 'j_sequence_start', 'j_sequence_end', 'c_sequence_start', 'c_sequence_end', 'consensus_count', 'duplicate_count', 'is_cell', 'locus', 'rearrangement_status', 'ambiguous'
metadata: 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'duplicate_count_B_VDJ', 'duplicate_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
Check the AnnData object as well
And the AnnData object is indexed based on cells.
[9]:
adata
[9]:
AnnData object with n_obs × n_vars = 10553 × 36601
obs: 'sample_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'scrublet_score', 'is_doublet', 'filter_rna', 'has_contig', 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'duplicate_count_B_VDJ', 'duplicate_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
var: 'gene_ids', 'feature_types', 'genome'
The number of cells that actually has a matching BCR can be tabluated.
[10]:
pd.crosstab(adata.obs["has_contig"], adata.obs["chain_status"])
[10]:
| chain_status | Extra pair | Extra pair-exception | No_contig | Orphan VDJ | Orphan VJ | Single pair |
|---|---|---|---|---|---|---|
| has_contig | ||||||
| No_contig | 0 | 0 | 9569 | 0 | 0 | 0 |
| True | 57 | 2 | 0 | 5 | 39 | 881 |
Now actually filter the AnnData object and run through a standard workflow starting by filtering genes and normalizing the data
Because the ‘filtered’ AnnData object was returned as a filtered but otherwise unprocessed object, we still need to normalize and run through the usual process here. The following is just a standard scanpy workflow.
[11]:
adata = adata[
adata.obs["filter_rna"] == "False"
] # from ddl.pp.recipe_scanpy_qc
# filter genes
sc.pp.filter_genes(adata, min_cells=3)
# Normalize the counts
sc.pp.normalize_total(adata, target_sum=1e4)
# Logarithmize the data
sc.pp.log1p(adata)
# Stash the normalised counts
adata.raw = adata
Identify highly-variable genes
[12]:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)
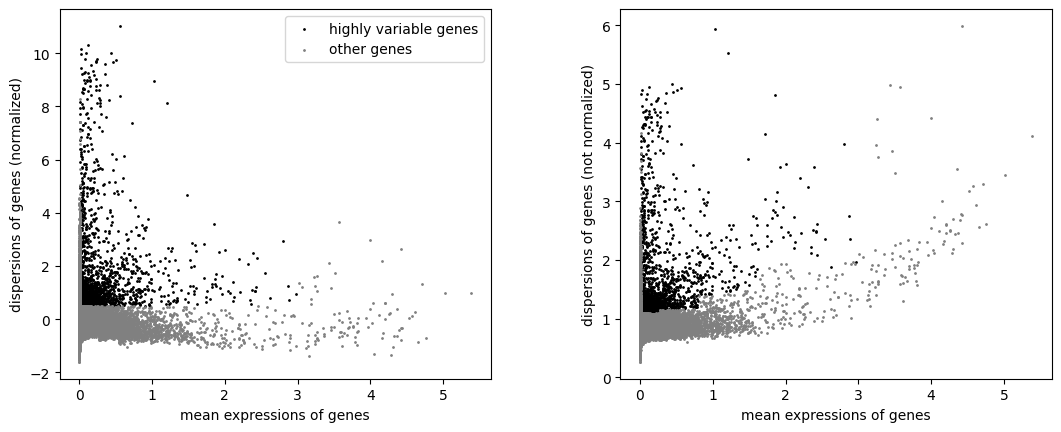
Filter the genes to only those marked as highly-variable
[13]:
adata = adata[:, adata.var.highly_variable]
Regress out effects of total counts per cell and the percentage of mitochondrial genes expressed. Scale the data to unit variance.
[14]:
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"])
sc.pp.scale(adata, max_value=10)
Run PCA
[15]:
sc.tl.pca(adata, svd_solver="arpack")
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=50)

Computing the neighborhood graph, umap and clusters
[16]:
# Computing the neighborhood graph
sc.pp.neighbors(adata)
# Embedding the neighborhood graph
sc.tl.umap(adata)
# Clustering the neighborhood graph
sc.tl.leiden(adata)
Visualizing the clusters and whether or not there’s a corresponding BCR
[17]:
sc.pl.umap(adata, color=["leiden", "chain_status"])

Visualizing some B cell genes
[18]:
sc.pl.umap(adata, color=["IGHM", "JCHAIN"])

Save AnnData
We can save this AnnData object for now.
[19]:
adata.write("adata2.h5ad", compression="gzip")
Save dandelion
To save the vdj object, we have two options - either save the .data and .metadata slots with pandas’ functions:
[20]:
vdj.data.to_csv("filtered_vdj_table2.tsv", sep="\t")
[21]:
vdj.write_h5ddl("dandelion_results2.h5ddl", complib="bzip2")
Concatenating multiple bcr objects
It is quite common that one might be trying to analyse data from multiple samples. In that case, dandelion has a concat function to merge the data.
We will simulate a second object but reading in the same file.
[22]:
vdj1 = ddl.read_10x_airr(file_location)
vdj2 = ddl.read_10x_airr(file_location)
Before you merge the objects, make sure that the “cell_id” and “sequence_id” are distinct so that you can distinguish them later
[23]:
# note: if the you reannotated the data with the full processing pipeline i.e. ran ddl.pp.format_fasta, this is already taken care of.
vdj1.data.cell_id = ["run1" + "_" + x for x in vdj1.data.cell_id]
vdj2.data.cell_id = ["run2" + "_" + x for x in vdj2.data.cell_id]
# probably don't need to modify the sequence_id. the concat function will check for unique and append -0, -1 etc when it encounters non-unique sequence_ids
# vdj1.data.sequence_id = ["run1" + '_' + x for x in vdj1.data.sequence_id]
# vdj2.data.sequence_id = ["run2" + '_' + x for x in vdj2.data.sequence_id]
# you can also reinitialise just in case so that the info is propagated properly (unnecessary here)
# vdj1 = ddl.Dandelion(vdj1.data)
# vdj2 = ddl.Dandelion(vdj2.data)
[24]:
vdj_merged = ddl.concat([vdj1, vdj2])
vdj_merged
[24]:
Dandelion class object with n_obs = 1988 and n_contigs = 4186
data: 'cell_id', 'sequence_id', 'sequence', 'sequence_aa', 'productive', 'rev_comp', 'v_call', 'v_cigar', 'd_call', 'd_cigar', 'j_call', 'j_cigar', 'c_call', 'c_cigar', 'sequence_alignment', 'germline_alignment', 'junction', 'junction_aa', 'junction_length', 'junction_aa_length', 'v_sequence_start', 'v_sequence_end', 'd_sequence_start', 'd_sequence_end', 'j_sequence_start', 'j_sequence_end', 'c_sequence_start', 'c_sequence_end', 'consensus_count', 'duplicate_count', 'is_cell', 'locus', 'rearrangement_status'
metadata: 'locus_VDJ', 'locus_VJ', 'productive_VDJ', 'productive_VJ', 'v_call_VDJ', 'd_call_VDJ', 'j_call_VDJ', 'v_call_VJ', 'j_call_VJ', 'c_call_VDJ', 'c_call_VJ', 'junction_VDJ', 'junction_VJ', 'junction_aa_VDJ', 'junction_aa_VJ', 'v_call_B_VDJ', 'd_call_B_VDJ', 'j_call_B_VDJ', 'v_call_B_VJ', 'j_call_B_VJ', 'c_call_B_VDJ', 'c_call_B_VJ', 'productive_B_VDJ', 'productive_B_VJ', 'duplicate_count_B_VDJ', 'duplicate_count_B_VJ', 'v_call_VDJ_main', 'v_call_VJ_main', 'd_call_VDJ_main', 'j_call_VDJ_main', 'j_call_VJ_main', 'c_call_VDJ_main', 'c_call_VJ_main', 'v_call_B_VDJ_main', 'd_call_B_VDJ_main', 'j_call_B_VDJ_main', 'v_call_B_VJ_main', 'j_call_B_VJ_main', 'isotype', 'isotype_status', 'locus_status', 'chain_status', 'rearrangement_status_VDJ', 'rearrangement_status_VJ'
[25]:
vdj_merged.data
[25]:
| cell_id | sequence_id | sequence | sequence_aa | productive | rev_comp | v_call | v_cigar | d_call | d_cigar | ... | d_sequence_end | j_sequence_start | j_sequence_end | c_sequence_start | c_sequence_end | consensus_count | duplicate_count | is_cell | locus | rearrangement_status | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequence_id | |||||||||||||||||||||
| AAACCTGTCATATCGG-1_contig_1-0 | run1_AAACCTGTCATATCGG-1 | AAACCTGTCATATCGG-1_contig_1-0 | TGGGGAGGAGTCAGTCCCAACCAGGACACGGCCTGGACATGAGGGT... | MRVPAQLLGLLLLWLSGARCDIQMTQSPSSLSASVGDRVTITCQAT... | T | F | IGKV1-8 | 38S314M204S | NaN | NaN | ... | NaN | 384 | 420 | 421 | 556 | 9139 | 68 | T | IGK | standard |
| AAACCTGTCCGTTGTC-1_contig_2-0 | run1_AAACCTGTCCGTTGTC-1 | AAACCTGTCCGTTGTC-1_contig_2-0 | ATCACATAACAACCACATTCCTCCTCTAAAGAAGCCCCTGGGAGCA... | MDWTWRFLFVVAAATGVQSQVQLVQSGAEVKKPGSSVKVSCKASGG... | T | F | IGHV1-69D | 58S353M154S | IGHD3-22 | 411S31M123S | ... | 442.0 | 445 | 494 | 495 | 565 | 4161 | 51 | T | IGH | standard |
| AAACCTGTCCGTTGTC-1_contig_1-0 | run1_AAACCTGTCCGTTGTC-1 | AAACCTGTCCGTTGTC-1_contig_1-0 | AGGAGTCAGACCCTGTCAGGACACAGCATAGACATGAGGGTCCCCG... | MRVPAQLLGLLLLWLPGARCAIRMTQSPSSFSASTGDRVTITCRAS... | T | F | IGKV1-8 | 33S345M173S | NaN | NaN | ... | NaN | 378 | 415 | 416 | 551 | 5679 | 43 | T | IGK | standard |
| AAACCTGTCGAGAACG-1_contig_1-0 | run1_AAACCTGTCGAGAACG-1 | AAACCTGTCGAGAACG-1_contig_1-0 | ACTGTGGGGGTAAGAGGTTGTGTCCACCATGGCCTGGACTCCTCTC... | MAWTPLLLLFLSHCTGSLSQAVLTQPSSLSASPGASGRLTCTLRSD... | T | F | IGLV5-45 | 28S369M245S | NaN | NaN | ... | NaN | 394 | 431 | 432 | 642 | 13160 | 90 | T | IGL | standard |
| AAACCTGTCGAGAACG-1_contig_2-0 | run1_AAACCTGTCGAGAACG-1 | AAACCTGTCGAGAACG-1_contig_2-0 | GGGAGCATCACCCAGCAACCACATCTGTCCTCTAGAGAATCCCCTG... | MDWTWRILFLVAAATGAHSQVQLVQSGGEVKKPGASVKVSCKASGY... | T | F | IGHV1-2 | 64S353M133S | NaN | NaN | ... | NaN | 430 | 479 | 480 | 550 | 5080 | 47 | T | IGH | standard |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TTTGGTTTCAGAGCTT-1_contig_2-1 | run2_TTTGGTTTCAGAGCTT-1 | TTTGGTTTCAGAGCTT-1_contig_2-1 | GGGAGAGCCCTGGGGAGGAACTGCTCAGTTAGGACCCAGAGGGAAC... | MEAPAQLLFLLLLWLPDTTGEIVLTQSPATLSLSPGERATLSCRAS... | T | F | IGKV3-11 | 47S345M170S | NaN | NaN | ... | NaN | 389 | 426 | 427 | 562 | 11867 | 73 | T | IGK | standard |
| TTTGGTTTCAGTGTTG-1_contig_1-1 | run2_TTTGGTTTCAGTGTTG-1 | TTTGGTTTCAGTGTTG-1_contig_1-1 | GGGGTCACAAGAGGCAGCGCTCTCGGGACGTCTCCACCATGGCCTG... | MAWALLLLTLLTQDTGSWAQSALTQPASVSGSPGQSITISCTGTSS... | T | F | IGLV2-23 | 38S340M262S | NaN | NaN | ... | NaN | 392 | 429 | 430 | 640 | 6497 | 58 | T | IGL | standard |
| TTTGGTTTCAGTGTTG-1_contig_2-1 | run2_TTTGGTTTCAGTGTTG-1 | TTTGGTTTCAGTGTTG-1_contig_2-1 | ATATTTCGTATCTGGGGAGTGACTCCTGTGCCCCACCATGGACACA... | MDTLCSTLLLLTIPSWVLSQITLKESGPTLVKPTQTLTLTCTFSGF... | T | F | IGHV2-5 | 37S358M122S | NaN | NaN | ... | NaN | 399 | 446 | 447 | 517 | 3530 | 33 | T | IGH | standard |
| TTTGGTTTCGGTGTCG-1_contig_2-1 | run2_TTTGGTTTCGGTGTCG-1 | TTTGGTTTCGGTGTCG-1_contig_2-1 | GGGAGAGCCCTGGGGAGGAACTGCTCAGTTAGGACCCAGAGGGAAC... | MEAPAQLLFLLLLWLPDTTGEIVLTQSPATLSLSPGERATLSCRAS... | T | F | IGKV3-11 | 47S345M176S | NaN | NaN | ... | NaN | 396 | 432 | 433 | 568 | 3058 | 22 | T | IGK | standard |
| TTTGGTTTCGGTGTCG-1_contig_1-1 | run2_TTTGGTTTCGGTGTCG-1 | TTTGGTTTCGGTGTCG-1_contig_1-1 | GAGAGAGGAGCCTTAGCCCTGGATTCCAAGGCCTATCCACTTGGTG... | MELGLRWVFLVAILEGVQCEVQLVESGGGLVKPGGSLRLSCAASGF... | T | F | IGHV3-21 | 73S353M145S | NaN | NaN | ... | NaN | 448 | 500 | 501 | 571 | 1026 | 12 | T | IGH | standard |
4186 rows × 33 columns
[26]:
vdj_merged.metadata
[26]:
| locus_VDJ | locus_VJ | productive_VDJ | productive_VJ | v_call_VDJ | d_call_VDJ | j_call_VDJ | v_call_VJ | j_call_VJ | c_call_VDJ | ... | d_call_B_VDJ_main | j_call_B_VDJ_main | v_call_B_VJ_main | j_call_B_VJ_main | isotype | isotype_status | locus_status | chain_status | rearrangement_status_VDJ | rearrangement_status_VJ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| run1_AAACCTGTCATATCGG-1 | None | IGK | None | T | None | None | None | IGKV1-8 | IGKJ4 | None | ... | None | None | IGKV1-8 | IGKJ4 | None | None | Orphan IGK | Orphan VJ | None | standard |
| run1_AAACCTGTCCGTTGTC-1 | IGH | IGK | T | T | IGHV1-69D | IGHD3-22 | IGHJ3 | IGKV1-8 | IGKJ1 | IGHM | ... | IGHD3-22 | IGHJ3 | IGKV1-8 | IGKJ1 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| run1_AAACCTGTCGAGAACG-1 | IGH | IGL | T | T | IGHV1-2 | None | IGHJ3 | IGLV5-45 | IGLJ3 | IGHM | ... | None | IGHJ3 | IGLV5-45 | IGLJ3 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
| run1_AAACCTGTCTTGAGAC-1 | IGH | IGK | T | T | IGHV5-51 | None | IGHJ3 | IGKV1D-8 | IGKJ2 | IGHM | ... | None | IGHJ3 | IGKV1D-8 | IGKJ2 | IgM | IgM | IGH + IGK | Single pair | standard | standard |
| run1_AAACGGGAGCGACGTA-1 | IGH | IGL | T | T | IGHV4-59 | None | IGHJ3 | IGLV3-19 | IGLJ2 | IGHM | ... | None | IGHJ3 | IGLV3-19 | IGLJ2 | IgM | IgM | IGH + IGL | Single pair | standard | standard |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| run2_ACGTCAAAGTTTCCTT-1 | IGH | None | T | None | IGHV3-21 | None | IGHJ4 | None | None | IGHM | ... | None | IGHJ4 | None | None | IgM | IgM | Orphan IGH | Orphan VDJ | standard | None |
| run2_CACTCCACAGATGGCA-1 | IGH | None | T | None | IGHV5-51 | None | IGHJ5 | None | None | IGHM | ... | None | IGHJ5 | None | None | IgM | IgM | Orphan IGH | Orphan VDJ | standard | None |
| run2_CGGTTAAGTTTCGCTC-1 | IGH | None | T | None | IGHV1-69D | None | IGHJ4 | None | None | IGHM | ... | None | IGHJ4 | None | None | IgM | IgM | Orphan IGH | Orphan VDJ | standard | None |
| run2_GTATCTTTCGAGAGCA-1 | IGH | None | T | None | IGHV3-23 | IGHD3-3 | IGHJ4 | None | None | IGHD | ... | IGHD3-3 | IGHJ4 | None | None | IgD | IgD | Orphan IGH | Orphan VDJ | standard | None |
| run2_TGACTTTGTTATCGGT-1 | IGH | None | T | None | IGHV1-69D | None | IGHJ3 | None | None | IGHM | ... | None | IGHJ3 | None | None | IgM | IgM | Orphan IGH | Orphan VDJ | standard | None |
1988 rows × 44 columns
[ ]: